Blog
March 12, 2026

Short answer: investors often interpret FDA Pre-Submission feedback as a signal of regulatory maturity and execution risk. A Pre-Submission (Pre-Sub) is designed to align a company with the U.S. Food and Drug Administration before a formal submission such as a 510(k), De Novo, or PMA.
March 11, 2026
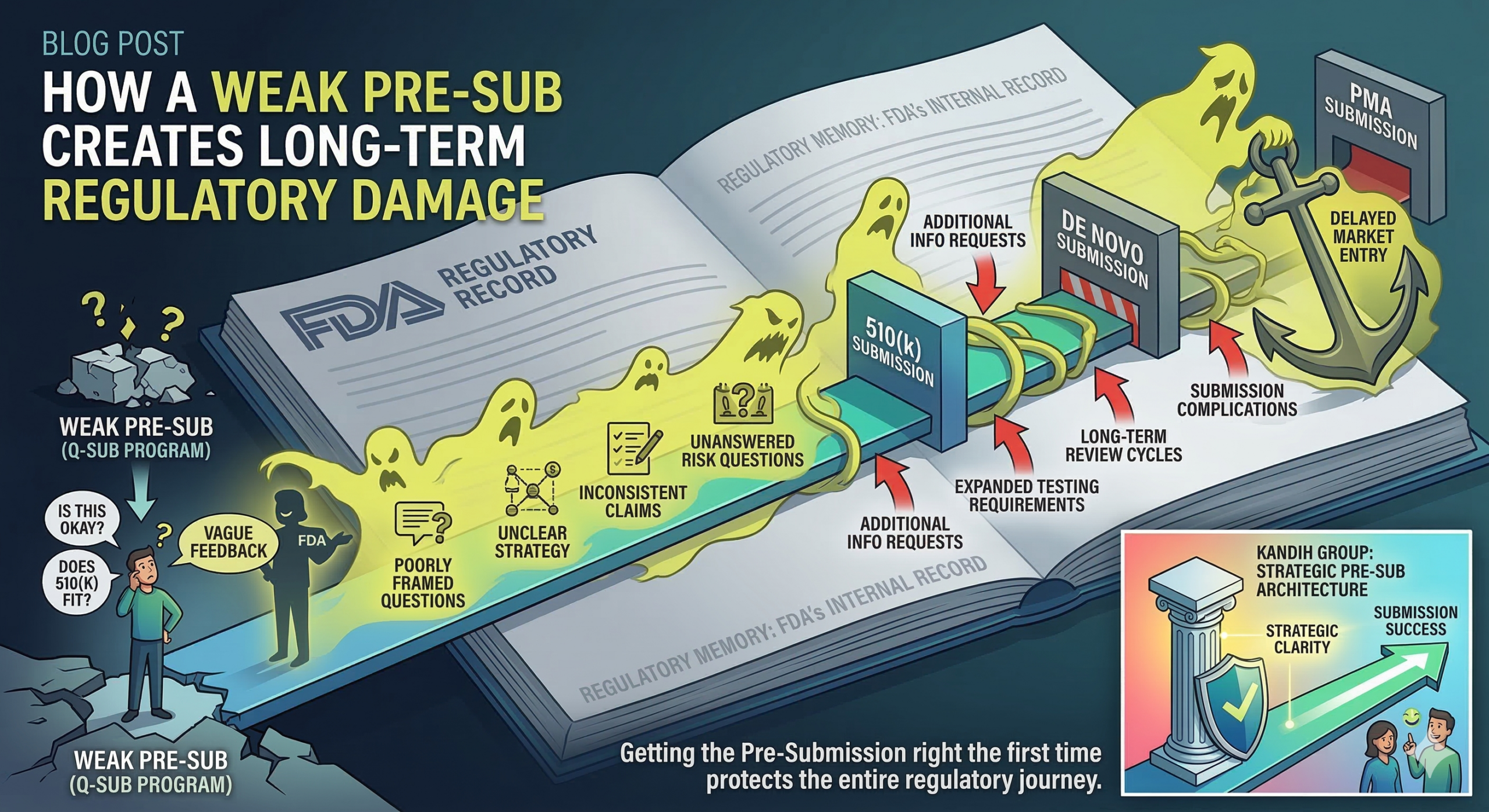
Short answer: a weak Pre-Submission (Pre-Sub) does not just waste a meeting with the FDA. It creates a regulatory record that can follow your device through the rest of development. Many founders treat a Pre-Sub as a casual conversation with the U.S. Food and Drug Administration. In reality, it is a formal interaction within the FDA’s Q-Submission program. The discussion becomes part of the agency’s internal regulatory history for your device.
March 10, 2026

Short answer: during a Pre-Submission (Pre-Sub), the FDA focuses on risk, regulatory pathway logic, and evidence strategy—not technical detail overload. Many founders assume the best way to impress reviewers at the U.S. Food and Drug Administration is to provide as much technical detail as possible.
March 9, 2026
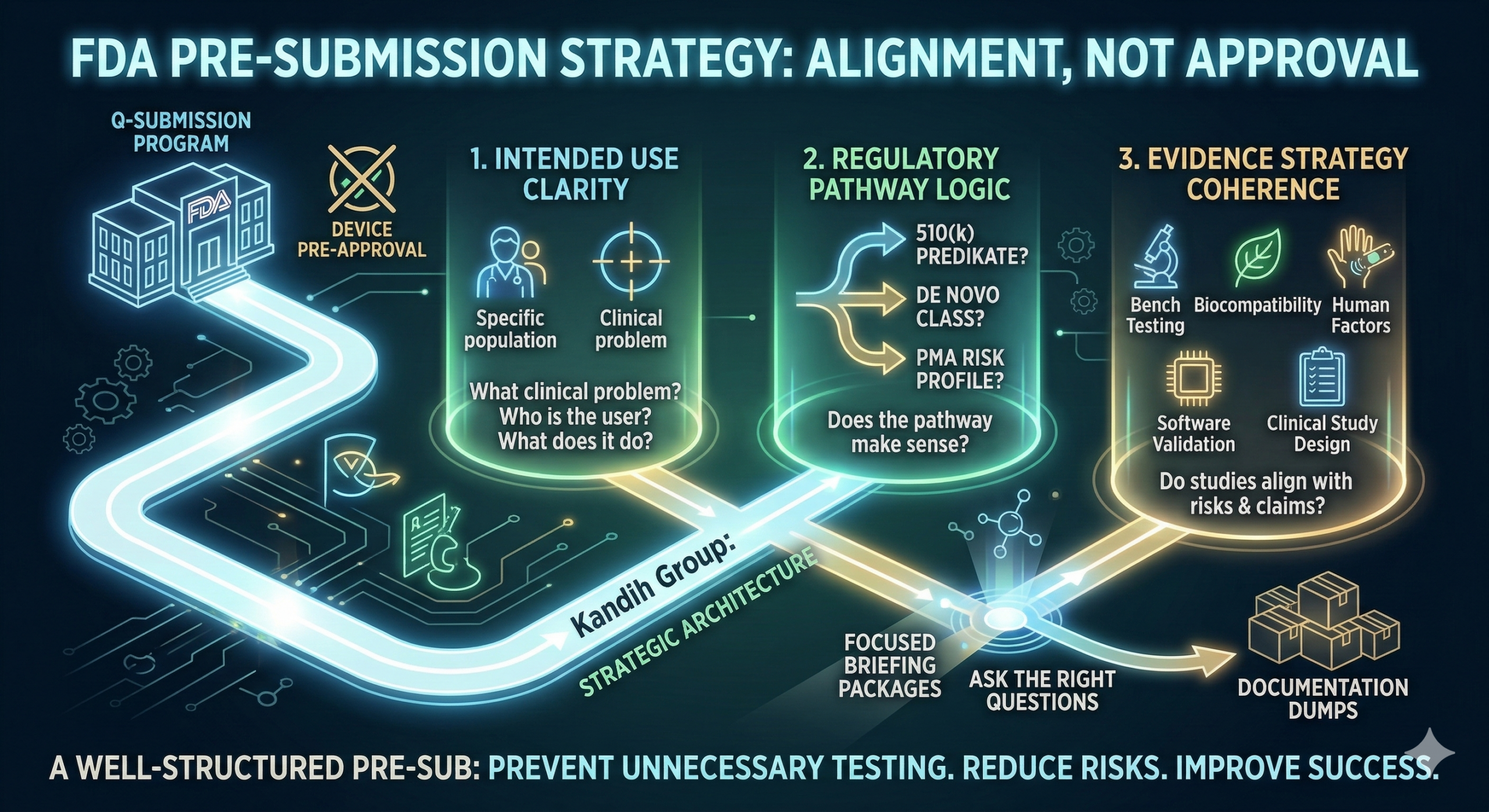
Short answer: the FDA expects a Pre-Submission (Pre-Sub) to clarify risk, regulatory pathway, and evidence strategy—not to pre-approve your device. A Pre-Submission meeting allows companies to ask the U.S. Food and Drug Administration targeted questions about their development plan before investing heavily in studies or submissions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}