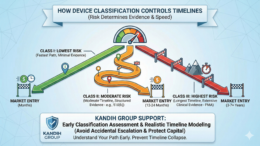
Straight answer: device classification controls how much evidence you must generate—and that controls how fast (or slow) you reach market.
If you don’t understand your classification early, your timeline is not a plan. It’s a guess.
Under the framework of the U.S. Food and Drug Administration, medical devices are placed into three risk-based classes: Class I, Class II, and Class III. The higher the risk, the heavier the evidence burden—and the longer the timeline.
Let’s break down what that means in real terms.
Class I: Lowest Risk, Fastest Path (Usually)
Class I devices present minimal potential for harm.
Examples include basic surgical instruments and non-powered devices with well-understood risks.
What This Means for Timelines:
Often exempt from premarket submission
May only require general controls and quality system compliance
Limited testing burden
Typical timeline impact:
Months, not years
Lower development cost
Faster revenue entry
But don’t assume your device is Class I just because it looks simple. Intended use can escalate classification quickly.
Class II: Moderate Risk, Moderate Timeline
Most devices fall into Class II.
These require demonstration of safety and effectiveness—usually through the 510(k) pathway.
What This Means for Timelines:
Bench testing is required
Risk-based biocompatibility may be required
Software validation if applicable
Human factors evaluation in many cases
FDA review cycles apply
Typical timeline impact:
12–24 months from concept to clearance (depending on readiness)
Moderate regulatory spend
Iterative FDA interaction possible
Class II is manageable—but only if evidence is aligned correctly from the start.
Class III: Highest Risk, Longest Timeline
Class III devices sustain or support life, are implanted long-term, or present significant risk if they fail.
These require Premarket Approval (PMA).
What This Means for Timelines:
Extensive bench testing
Often animal studies
Well-controlled clinical trials
Comprehensive benefit-risk analysis
Longer FDA review periods
Typical timeline impact:
3–7+ years
Multi-million-dollar evidence programs
Significant investor dependence
At this level, classification doesn’t just affect timeline—it defines your capital strategy.
Why Classification Changes Everything
Device classification determines:
Type of submission required
Amount of preclinical testing
Need for clinical trials
Review timelines
Regulatory cost structure
Investor risk profile
If your classification is wrong, your timeline model is wrong.
And if your timeline model is wrong, your capital runway may be wrong too.
That’s how companies unexpectedly run out of money mid-development.
The Hidden Risk: Accidental Escalation
Common early decisions that escalate classification:
Expanding intended use claims
Targeting vulnerable populations
Adding therapeutic functionality
Incorporating novel materials or AI-based decision support
These changes may feel small technically.
Regulatorily, they can shift you from Class II to Class III.
That is not a minor adjustment. It is a strategic reset.
Where Kandih Comes In
This is where Kandih Group provides early clarity.
Kandih conducts structured classification assessments that:
Analyze intended use against FDA regulatory codes
Evaluate predicate device comparability
Identify technological risk escalators
Map classification to required evidence categories
Model realistic development timelines tied to risk level
Align timeline projections with capital planning
Instead of assuming a timeline, teams build one based on regulatory reality.
That prevents:
Forced pathway changes
Investor misalignment
Unexpected testing programs
Timeline collapse under diligence
Bottom Line
Device classification is not just a regulatory label.
It is a timeline controller.
Class I moves fastest.
Class II requires structured evidence.
Class III demands long-term capital and clinical proof.
Understanding your classification early protects time, money, and credibility.
That’s how Kandih helps founders build realistic, defensible paths to market—before engineering decisions harden.
References
FDA – Classify Your Medical Device
https://www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device
FDA – Premarket Notification 510(k)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-notification-510k
FDA – Premarket Approval (PMA)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma
FDA – De Novo Classification Process
https://www.fda.gov/medical-devices/premarket-submissions/de-novo-classification-request