Silent Killers of 510(k) Submissions: Biocompatibility Report Gaps
By Kandih Bioscience • Regulatory Strategy Series
The truth is, it rarely comes out of nowhere. There are specific, well-documented gaps in biocompatibility reports that trigger FDA deficiencies almost every single time. They’re not obvious. They’re not always covered in training. And they can quietly kill a submission that should have sailed through.
Let’s walk through what they are — and how to make sure your submission doesn’t fall into the same traps.
First, a Quick Reality Check on eSTAR
There’s a myth floating around that if your eSTAR submission validates without errors, you’re good to go. The file goes through, the system accepts it, and FDA starts the clock.
Here’s the problem: validation only checks the format of your submission. It checks that the right boxes are filled in, that the right documents are attached, and that the technical structure of your file is correct.
It does not check whether the content inside those documents actually answers FDA’s questions.
A biocompatibility report can be perfectly formatted and completely inadequate at the same time. And that gap — between a technically valid submission and a scientifically complete one — is exactly where most 510(k) biocompatibility deficiencies live.
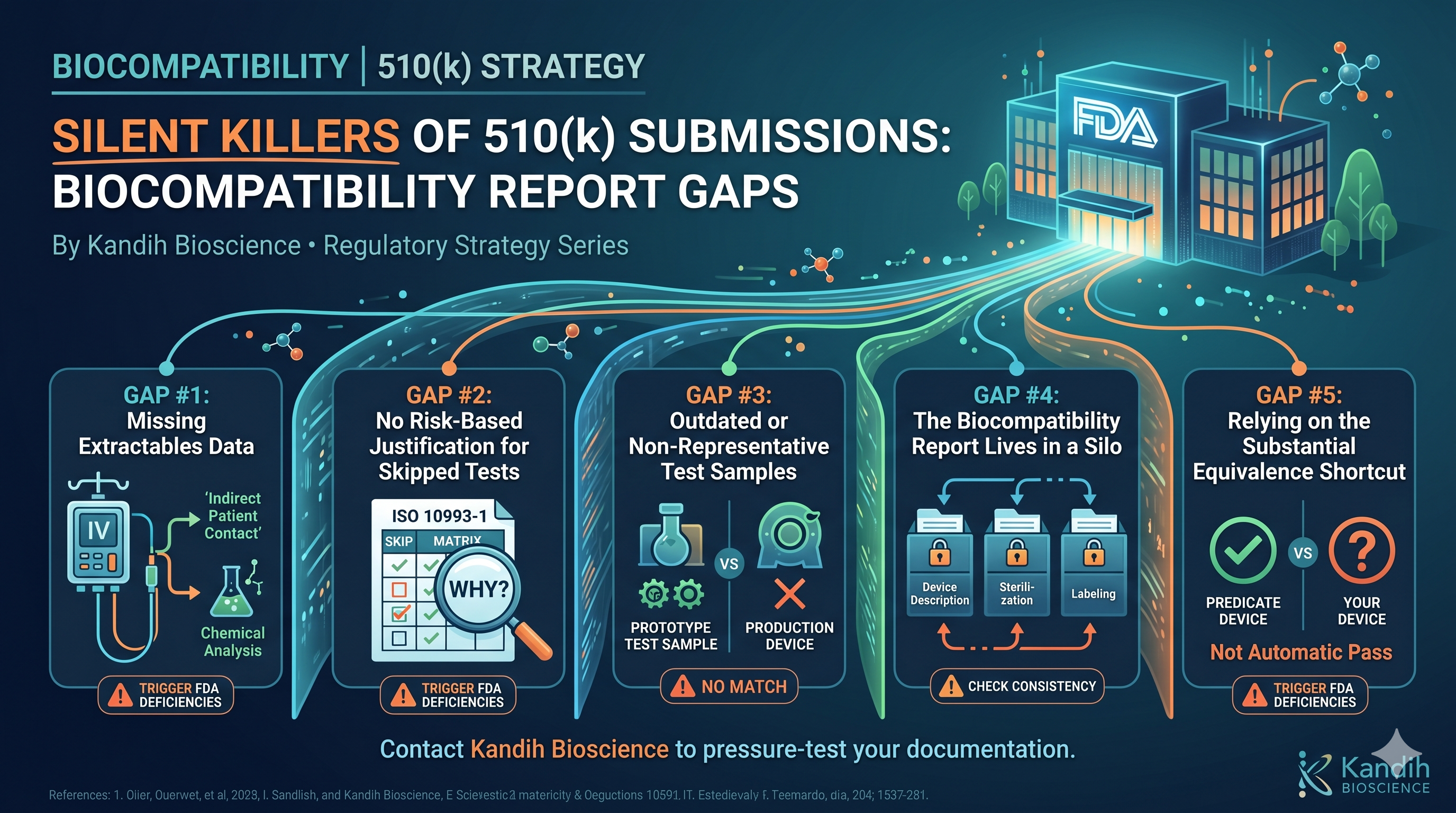
Gap #1: Missing Extractables Data for Indirect Contact Devices
A lot of device manufacturers focus their biocompatibility testing on the parts of their device that directly touch the patient. That makes sense. But they forget about the parts that indirectly interact with the patient — specifically, through fluid pathways.
Think about a device that connects to an IV line, or a pump that moves fluid that will eventually enter a patient’s bloodstream. The device itself might never touch the patient. But if chemicals can leach from the device into that fluid, those chemicals absolutely can reach the patient.
Under ISO 10993-18, you’re required to characterize those leachable chemicals through an extractables study. A lot of submissions either skip this entirely or include a brief statement saying the materials are “biocompatible” without any supporting chemistry data.
FDA does not accept material certifications or supplier statements as a substitute for extractables data when indirect contact with fluids is involved. If you’re not conducting the chemistry, you’re likely getting a deficiency letter.
Gap #2: No Risk-Based Justification for Skipped Tests
ISO 10993-1 does not require you to run every single biocompatibility test. It requires you to think critically about which tests are relevant for your specific device — based on the contact type, contact duration, and the nature of the materials involved.
What trips up a lot of submissions is not that they skip tests. It’s that they skip tests without explaining why.
If you decide that a cytotoxicity study isn’t necessary for your device, you need to document your reasoning in a way that shows you actually thought it through. The same goes for genotoxicity, sensitization, systemic toxicity, and every other endpoint in the standard.
FDA reviewers are not going to give you the benefit of the doubt. If a test is missing and there’s no documented justification, the default assumption is that it was overlooked — not strategically excluded. And that’s an automatic deficiency.
The fix is simple: your biological evaluation plan needs to walk through each test endpoint, state whether testing was conducted or waived, and explain the scientific reasoning behind the decision. No gaps, no assumptions.
Gap #3: Outdated or Non-Representative Test Samples
This one catches people off guard because it feels like a procurement or logistics problem, not a regulatory one. But FDA cares deeply about it.
Your biocompatibility testing is only valid if the samples you tested are representative of the device that’s going to market. That means the same materials, the same manufacturing process, the same colorants, the same coatings, and the same sterilization method.
Here’s where companies run into trouble:
- Testing was done on prototype materials, but the production version uses a slightly different polymer formulation
- A colorant or pigment was changed after testing was completed, without re-evaluating the biological risk
- The device was tested before final sterilization validation, so the testing didn’t account for the chemical changes that sterilization can cause
- A secondary supplier was added for a critical component, but no new testing was triggered
Any one of these scenarios can invalidate your existing biocompatibility data. FDA will ask whether your test samples match your production device. If the answer is no — or if you can’t document that the answer is yes — you have a problem.
Gap #4: The Biocompatibility Report Lives in a Silo
Your 510(k) submission is a package. And every piece of that package needs to tell a consistent story.
What FDA reviewers notice is when the biocompatibility section says one thing and another part of the submission says something different. For example:
- The biocompatibility report references a material that doesn’t appear in your device description
- Your sterilization section describes a different sterilization method than the one documented in your bio testing
- Your labeling describes contact with a body surface that your biocompatibility section didn’t account for
These inconsistencies don’t always mean something is wrong with your device. But they signal to FDA that your documentation wasn’t carefully reviewed before submission. And once FDA starts pulling on that thread, they tend to pull hard.
The solution is a cross-functional review before you submit — someone needs to read the biocompatibility section alongside the device description, the manufacturing section, and the labeling, and confirm that they all line up.
Gap #5: Relying on the Substantial Equivalence Shortcut
If you’re submitting a 510(k), you’ve identified a predicate device — a device already on the market that your device is substantially equivalent to. This is the foundation of the 510(k) pathway.
But here’s what a lot of people misunderstand: substantial equivalence is a regulatory determination, not a biocompatibility determination.
Just because your predicate device had a certain biocompatibility profile doesn’t mean your device automatically inherits that profile. Your device has its own materials, its own manufacturing history, its own sterilization process. You need to evaluate it on its own merits.
Submissions that say, in effect, “our predicate passed biocompatibility, so we should too” are missing the point. FDA expects you to demonstrate — with actual data or documented scientific reasoning — that your device is safe for patient contact. The predicate is a baseline, not a pass.
Where Kandih Bioscience Comes In
Most of the deficiencies described above don’t happen because companies don’t care about quality. They happen because the regulatory requirements around biocompatibility are genuinely complex — and because the stakes are high enough that a small gap in documentation can cost months.
At Kandih Bioscience, our work starts before your submission does. We review your device’s material profile, identify which tests you actually need (and which you can justify skipping), evaluate your existing data for completeness, and help you build a biological evaluation report that is designed to answer FDA’s questions before they ask them.
We’ve helped clients navigate biocompatibility strategy for Class I, Class II, and Class III devices — across 510(k), De Novo, and PMA pathways. We know where the gaps tend to show up, and we know how to close them.
If you’re preparing a 510(k) submission and you want someone to pressure-test your biocompatibility documentation before it goes to FDA, that’s exactly what we do.
Contact Kandih Bioscience
References
1. FDA — Use of International Standard ISO 10993-1: Biological Evaluation of Medical Devices — Part 1 (2020)
2. FDA — eSTAR 510(k) Submission Template and Guidance (2023)
3. ISO 10993-18: Biological Evaluation of Medical Devices — Chemical Characterization of Medical Device Materials (2020)
4. FDA — Biocompatibility Testing of Medical Devices: Common Deficiencies (2021)
5. FDA — Deciding When to Submit a 510(k) for a Change to an Existing Device (2017)
6. ISO 10993-1: Biological Evaluation — Evaluation and Testing Within a Risk Management Process (2018)