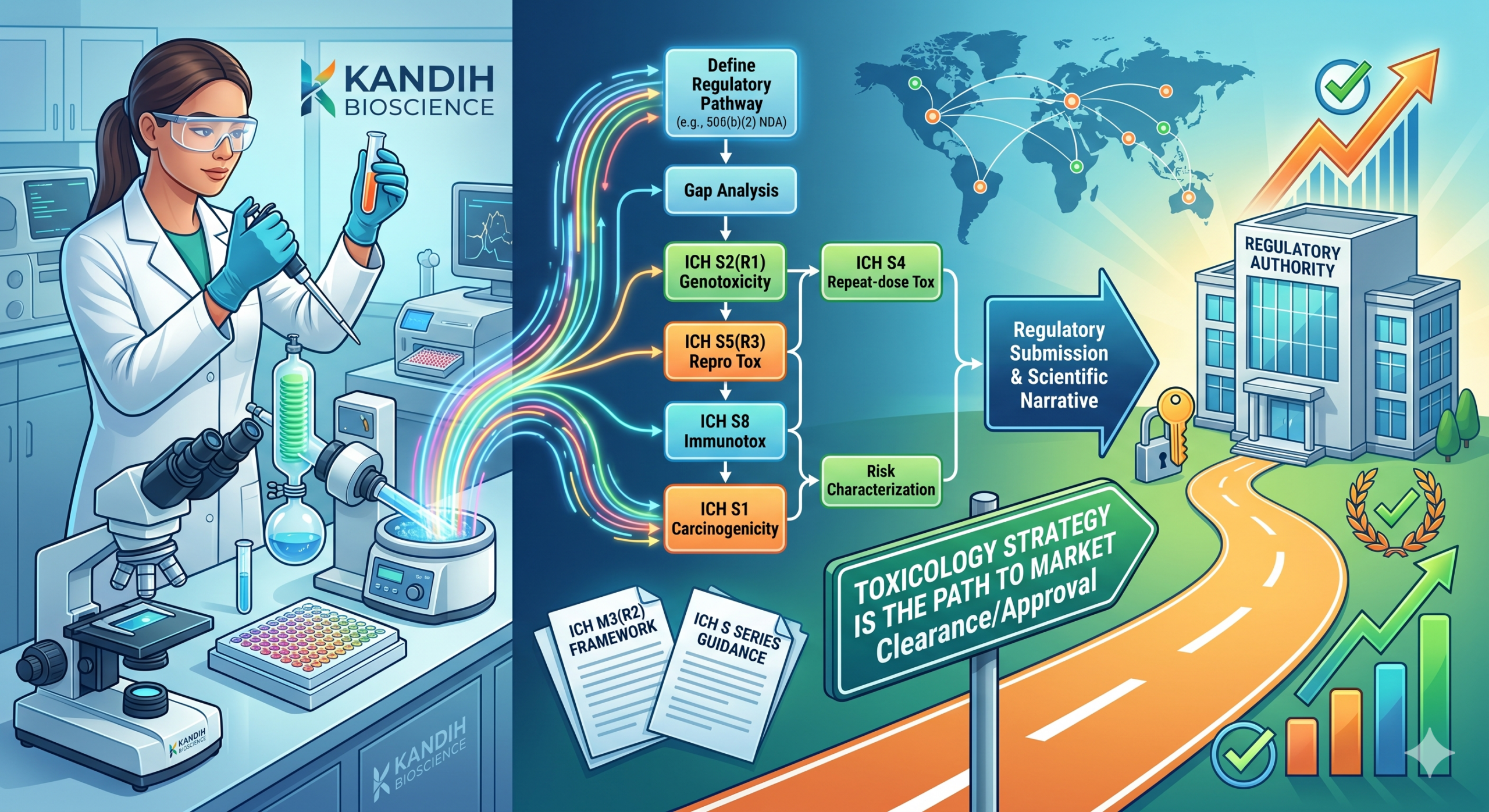
A toxicology strategy is a structured plan that connects your safety data to your regulatory pathway. It does more than confirm that a product won’t harm patients — it tells reviewers how you evaluated risk, why your evidence is sufficient, and what that means for market clearance or approval. Without it, even a clean set of tox studies can stall a submission.
If you are a founder or product developer bringing a pharmaceutical drug, biological treatment, or combination product to market, toxicology probably feels overwhelming. The science is dense. The regulatory expectations are moving targets. And the cost of getting it wrong — a Complete Response Letter, a study repeat, a delayed launch — is enormous.
Here is the problem most teams run into: they focus on running toxicology studies without building a strategy around them. Those are two very different things, and confusing them is one of the most common — and most expensive — mistakes in drug and device development.
What Is a Toxicology Strategy?
A toxicology strategy is a forward-looking plan that defines which safety assessments you need, in what order, and how each one feeds into your regulatory submission. It is not a list of tests. It is a scientific argument that your product is safe for its intended use — one grounded in ICH M3(R2), the globally recognized framework for nonclinical safety studies supporting human clinical trials and marketing authorization.
A well-built strategy does three things:
- Guides your product development decisions — which materials to use, which formulations to advance, which manufacturing changes require re-evaluation
- Shapes your regulatory submissions — whether you are filing a 510(k), an NDA, a BLA, or a combination product application, reviewers want to see that your safety evidence was collected systematically and interpreted against current guidelines
- Reduces downstream risk — catching a toxicological red flag in early development is a course correction; catching it during FDA review is a crisis
Toxicology studies are the inputs to that strategy. The strategy is what gives those inputs meaning.
Why Tox Studies Alone Don’t Guarantee Regulatory Success
This is a point that does not get said clearly enough: toxicology studies alone do not guarantee safety or regulatory success.
That sounds counterintuitive. If a product passes preclinical safety testing, why wouldn’t that be sufficient?
Because regulators are not just asking “is it safe?” They are asking “did you demonstrate safety in a way that is scientifically defensible, appropriately scoped, and aligned with the regulatory standard for this product class?” Those are harder questions, and they require more than a stack of clean study reports.
Common gaps that get products into trouble:
- Wrong study scope. A study may be conducted at doses, durations, or in animal models that do not reflect the intended clinical use. FDA’s guidance on nonclinical studies is explicit that the adequacy of a study is evaluated in the context of the intended clinical program — not in isolation.
- Missing genotoxicity or carcinogenicity data. ICH S2(R1) defines the standard battery for genotoxicity assessment. Missing a required endpoint — even unintentionally — triggers deficiency letters.
- Inadequate risk characterization. For combination products and biologics especially, FDA and other health authorities want to see that you understand the interaction between your device or drug component and the biological environment. ICH S8 provides the framework for immunotoxicology risk assessment that reviewers expect for biologics. A study result is not the same as a risk characterization.
- No narrative connecting the data. A regulatory submission is a scientific argument, not a document dump. If your tox data is not organized around a clear safety narrative, reviewers will ask for one — at the cost of months.
The teams that navigate this well are the ones that built a strategy before they ran the studies, not after.
What a Toxicology Risk Assessment Actually Covers
For pharmaceutical drugs, biologics, and combination products, a toxicology risk assessment is the formal analysis that sits at the center of your safety case. It evaluates potential hazards from your product’s chemical composition, biological interactions, route of administration, dose, and patient population.
Depending on your product type, this typically includes:
- General toxicology — acute, subchronic, and chronic studies conducted per ICH S4 in relevant animal models
- Genotoxicity — the standard battery defined under ICH S2(R1): Ames test, in vitro micronucleus, in vivo follow-up if needed
- Reproductive and developmental toxicity — required for products used in patients of childbearing potential, per ICH S5(R3)
- Carcinogenicity — assessed using the ICH S1 guideline series for products intended for chronic administration
- Immunotoxicology — particularly relevant for biologics and immunosuppressive therapies; evaluated per ICH S8
- Extractables and leachables — for combination products where a device component contacts drug or patient tissue, per FDA guidance on container closure systems
Each of these feeds into the risk characterization that regulators use to determine whether the benefit-risk profile supports market authorization.
The Gap Between Lab Testing and Government Approval
Here is where many teams underestimate the complexity: lab testing and regulatory approval are not directly connected. There is a significant interpretive layer in between — and that layer requires specialized expertise.
The question is not just whether your product is safe. It is whether your evidence of safety meets the standard for the specific regulatory pathway you are pursuing. A 505(b)(2) NDA, for example, has different nonclinical data expectations than a full 505(b)(1), and a De Novo carries different requirements than a PMA. Answering that question requires someone who understands both the science and the regulatory framework — someone who can translate a toxicology dataset into a submission-ready argument that holds up under review.
This is exactly where toxicology consultants like Kandih Bioscience add the most value. Their work is not limited to running or overseeing studies. It includes supporting regulatory submissions, reviewing the strategic adequacy of your safety data before you file, identifying gaps that would otherwise surface as reviewer deficiencies, and ensuring that your tox package tells a coherent scientific story.
For pharmaceutical drugs, biological treatments, and combination products — all of which carry complex, product-specific regulatory expectations under 21 CFR Part 3 — that interpretive expertise is the difference between a submission that advances and one that stalls.
How to Build a Toxicology Strategy That Works
The earlier you engage, the better. Toxicology strategy is not a pre-submission checklist item — it is a development-phase decision that affects study design, timeline, and budget.
Here is what a practical toxicology strategy process looks like:
- Define your regulatory pathway first. The required tox package for a 505(b)(2) NDA is different from a full 505(b)(1). A De Novo for a combination product has different expectations than a PMA. Your strategy has to be anchored to your intended pathway.
- Map your data gaps. What do you have? What does the pathway require? Where are the differences? That gap analysis drives your study plan. FDA’s Nonclinical Safety Review Guidance is a useful reference for understanding what reviewers will look for.
- Sequence studies to support development decisions. Early genotoxicity data can inform formulation choices. Repeat-dose tox can support dose selection for Phase 1 per ICH M3(R2). Build the strategy around decision gates, not just regulatory submission requirements.
- Prepare your risk characterization in parallel. Don’t wait until the study reports arrive to start writing the safety narrative. Draft the framework early so the final report data can slot in cleanly.
- Work with experts who understand both science and regulation. Toxicology consulting firms that specialize in your product type — particularly those with experience supporting FDA submissions — can compress timelines significantly by getting the strategy right before any studies are run.
Frequently Asked Questions
What is the difference between a toxicology study and a toxicology strategy? A toxicology study generates safety data for a specific endpoint — for example, genotoxicity per ICH S2(R1) or repeat-dose toxicity per ICH S4. A toxicology strategy determines which studies are needed, in what order, and how the resulting data will be used to build your regulatory safety case.
Do I need a toxicology strategy if my product is based on an established active ingredient? Yes. Even with prior human safety data, the specific formulation, delivery mechanism, dose, duration, and patient population of your product may introduce new toxicological considerations. FDA’s guidance on 505(b)(2) applications explains how prior data can be leveraged — but also where gaps still need to be addressed. A strategy helps you determine what new data is required.
When should I start building a toxicology strategy? Ideally during early development, before study designs are finalized. ICH M3(R2) specifically frames nonclinical studies as tools to support clinical development decisions — which is only possible if the strategy is built before the studies are run.
What types of products require the most complex toxicology risk assessments? Combination products (drug-device, biologic-device), novel biologics, products with new excipients or novel manufacturing processes, and products intended for vulnerable populations (pediatrics per FDA’s pediatric safety guidance, immunocompromised patients) typically require the most comprehensive toxicology evaluation.
How does toxicology consulting support regulatory submissions? Toxicology consultants review your data package for completeness and regulatory adequacy, write or review the toxicology sections of your submission, respond to reviewer questions, and advise on study design to fill identified gaps — all in the context of your specific FDA regulatory pathway.
Don’t Just Run Tox Studies. Build a Strategy.
If your product development plan treats toxicology as a checkbox — run the studies, include the reports, move on — you are taking on significant risk. Regulatory reviewers are looking for a coherent safety argument built around the right data. That takes strategy, not just studies.
Working with a toxicology consulting firm that specializes in your product type gives you more than scientific expertise. It gives you a team that understands how safety data translates into regulatory outcomes — and can help you build the strongest possible case before you file.
Ready to build a toxicology strategy for your product? Contact Kandih Bioscience at info@kandih.com or call 240.565.8933 to schedule a consultation.
Kandih Bioscience specializes in toxicology risk assessments and regulatory consulting for pharmaceutical drugs, biological treatments, and combination products. Their experts support regulatory submissions and oversee research studies across the product development lifecycle.
References
- U.S. Food and Drug Administration. Guidances for Industry: Pharmacology/Toxicology. FDA Center for Drug Evaluation and Research (CDER). https://www.fda.gov/drugs/guidance-compliance-regulatory-information/guidances-drugs
- ICH Guidance for Industry. M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals. FDA CDER/CBER. January 2010. https://www.fda.gov/media/71542/download
- ICH Guidance for Industry. S2(R1) Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use. FDA CDER/CBER. June 2012. https://www.federalregister.gov/documents/2012/06/07/2012-13774/international-conference-on-harmonisation-guidance-on-s2r1-genotoxicity-testing-and-data
- ICH Guidance for Industry. S8 Immunotoxicity Studies for Human Pharmaceuticals. FDA CDER/CBER. April 2006. https://www.fda.gov/media/72047/download
- U.S. Food and Drug Administration. Nonclinical Safety Evaluation of Reformulated Drug Products and Products Intended for Administration by an Alternate Route. FDA CDER. https://www.fda.gov/files/drugs/published/Nonclinical-Safety-Evaluation-of-Reformulated-Drug-Products-and-Products-Intended-for-Administration-by-an-Alternate-Route.pdf
- U.S. Food and Drug Administration. Guidance for Industry and FDA Staff: Considerations for the Design, Development, and Analytical Procedures for Combination Products. Office of Combination Products (OCP). https://www.fda.gov/media/75273/download
- ICH Guidance for Industry. M3(R2) Nonclinical Safety Studies — Questions and Answers (R2). FDA CDER/CBER. February 2013. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/m3r2nonclinical-safety-studies-conduct-human-clinical-trials-and-marketing-authorization