The deal closed.
Capital transferred. Board seats assigned. Milestones locked in. Everyone moved on.
Then, six months later, a quiet update arrived in the investor’s inbox:
“FDA has requested additional data.”
Timelines shifted. Burn rate climbed. The next round became a conversation nobody wanted to have. The investment wasn’t struggling because of the product — it was struggling because regulatory risk had been left unattended after the term sheet was signed.
This is more common than most investors realize.
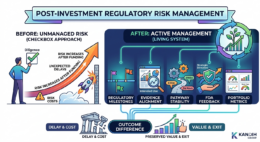
Regulatory Risk Doesn’t End at Closing — It Evolves
Most investors treat regulatory diligence as a box to check before wiring funds. Review the 510(k) strategy, confirm the predicate, assess the timeline — done.
But FDA doesn’t freeze the moment you close a round. Regulatory expectations shift as evidence develops, device designs change, product claims sharpen, and formal FDA interactions begin. What looked like a clean pathway at diligence can look very different twelve months into development.
Regulatory strategy is a living system. It requires active management, not a one-time review.
Why Risk Actually Increases After Funding
Counterintuitively, the moment a company receives investment is the moment regulatory exposure starts to grow.
Post-funding, everything accelerates — engineering, clinical work, claims development, timelines. That acceleration is the point. But it also means that small misalignments between what the company is building and what FDA expects can compound quietly, under the surface, until they surface as a formal request or a failed submission.
The assumptions made at diligence get tested. Evidence gaps become visible. FDA interactions become more formal and more consequential.
Without structured oversight, investors often don’t see these dynamics until the damage is already done.
Five Ways Investors Can Actively Manage Regulatory Risk
1. Add regulatory milestones alongside product milestones.
Most investor dashboards track engineering progress, market expansion, and revenue. But regulatory milestones — Pre-Submission completion, FDA feedback alignment, evidence plan finalization, submission readiness — are just as predictive of outcome. Tracking them creates accountability and surfaces delays early.
2. Ask whether data is answering the right questions, not just whether studies are running.
“Are studies progressing?” is the wrong question. The right question is: “Are these studies generating evidence that actually supports the intended use and satisfies the risk analysis?” Misaligned data leads to rework, not regulatory progress.
3. Reassess pathway stability on a regular cadence.
Regulatory pathways shift when designs change, new risks emerge, or FDA feedback reframes the evidence requirements. Investors should periodically ask: Is the original pathway still valid? Has anything changed the risk profile? Are contingency plans still relevant? Early detection of pathway instability is far less costly than late discovery.
4. Treat FDA feedback as strategic direction, not administrative correspondence.
FDA feedback isn’t informational — it’s directional. When FDA signals a concern or requests additional data, that signal needs to be clearly interpreted and translated into concrete changes to the development plan. Misreading FDA feedback is one of the most consistent sources of preventable delay in the industry.
5. Track regulatory risk as a living portfolio metric.
Regulatory risk belongs on the same dashboard as burn rate and runway. That means tracking the probability of a clinical requirement, the likelihood of additional studies, timeline variability, and open evidence gaps. What gets measured gets managed.
The Distinction That Changes Outcomes
Most investors treat regulatory strategy as a diligence checkpoint.
The strongest investors treat it as a portfolio management function — something that requires the same ongoing attention as financial performance, team dynamics, or market timing.
That shift in posture is where outcomes diverge.
Where Kandih Group Comes In
Kandih Group works with investors and medical device companies as an ongoing regulatory risk advisor — not just at the diligence stage, but throughout the development lifecycle.
That means continuously monitoring pathway stability, evaluating alignment between risk analysis, evidence, and claims, interpreting FDA feedback in real time, and translating regulatory developments into insights that are relevant to investment decisions.
The goal is simple: instead of reacting to regulatory problems when they surface, investors stay ahead of them.
The Real Lesson
The investor at the start of this story didn’t make a bad investment. They made an unmanaged one.
Regulatory risk doesn’t disappear after funding. It evolves — and left unmanaged, it compounds. Investors who build active regulatory oversight into their portfolio management protect timelines, preserve capital, and improve exit outcomes.
Those who treat it as a one-time checkpoint discover the risk late — when it’s hardest and most expensive to fix.
References
FDA – Requests for Feedback and Meetings for Medical Device Submissions (Q-Submission Program)
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/requests-feedback-and-meetings-medical-device-submissions-q-submission-program
FDA – Premarket Notification 510(k)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-notification-510k
FDA – De Novo Classification Process
https://www.fda.gov/medical-devices/premarket-submissions/de-novo-classification-request
FDA – Premarket Approval (PMA)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma