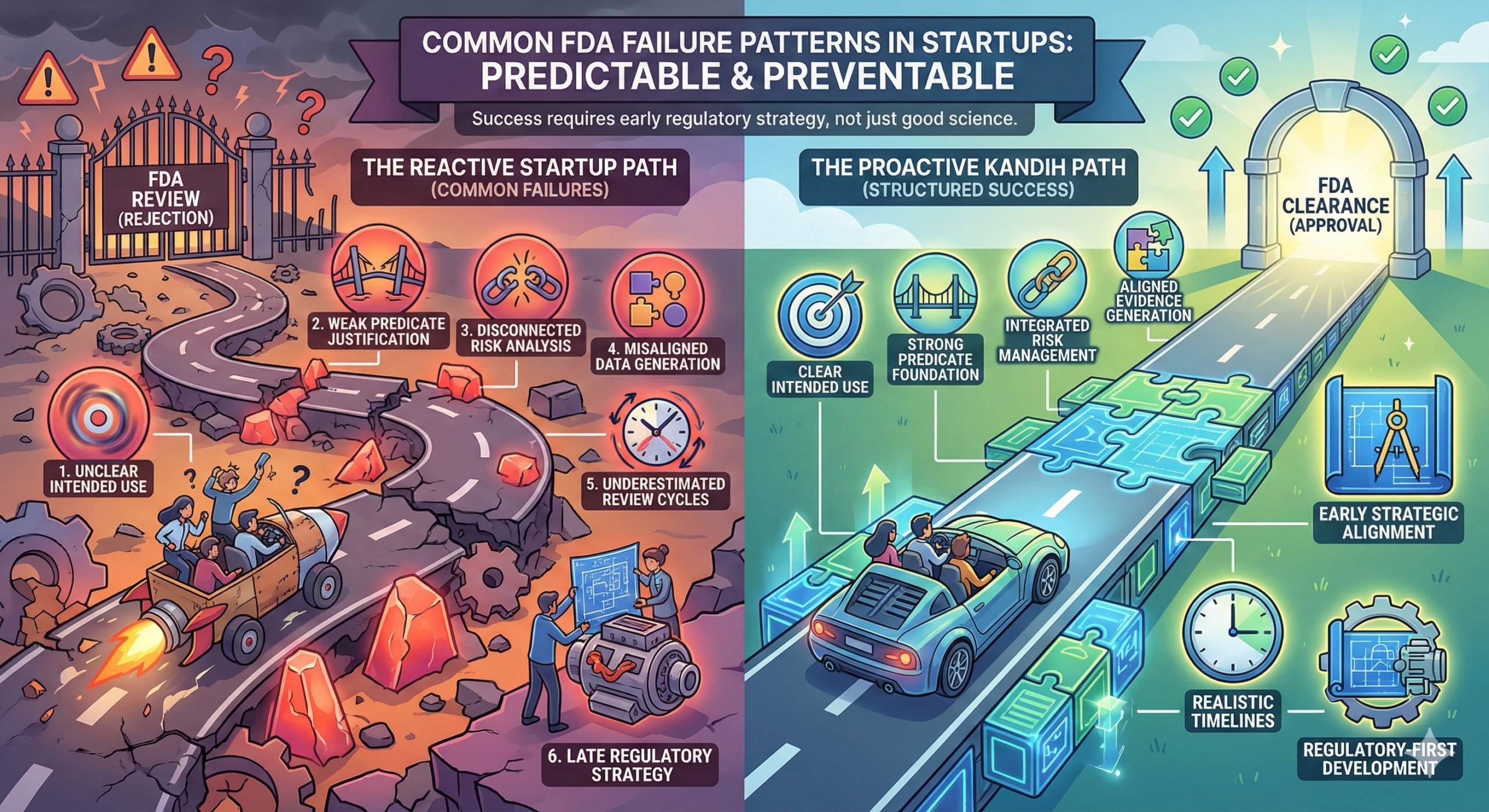
Short answer: most FDA submission failures follow predictable patterns. They are rarely random. They are rarely mysterious. And they are almost always preventable.
Startups often assume failure happens because the science was weak. In reality, failure usually happens because the regulatory strategy was fragmented, optimistic, or reactive.
Under the framework of the U.S. Food and Drug Administration, submissions are evaluated through structured risk, classification, and evidence logic. When a startup’s development plan does not align with that structure, problems surface quickly.
Here are the recurring failure patterns seen across early-stage companies.
1. Unclear or Overreaching Intended Use
This is one of the most common breakdowns.
Mistakes include:
Expanding claims beyond what evidence supports
Blurring diagnostic and therapeutic language
Targeting broader populations late in development
Using marketing language inside regulatory documents
If intended use shifts, the entire pathway may shift.
Why this fails: FDA evaluates the device based on intended use. If it is inconsistent or overly ambitious, evidence no longer matches the claim.
2. Weak Predicate Justification (510(k) Programs)
For companies pursuing 510(k), common issues include:
Selecting predicates based on similarity in marketing, not risk
Ignoring technological differences
Underestimating “new questions of safety or effectiveness”
Providing superficial comparison tables
If substantial equivalence is weak, the review becomes unstable.
3. Risk Analysis That Doesn’t Connect to Testing
FDA expects traceability:
Identified hazard
Risk control
Verification testing
Validation outcome
Startups often:
Perform risk analysis late
Treat it as documentation instead of strategy
Fail to align testing plans with identified hazards
Disconnected risk and evidence triggers additional information requests.
4. Generating Data Without Regulatory Alignment
Another recurring pattern:
Studies launched before pathway confirmation
Bench testing that does not map to safety endpoints
Clinical endpoints that do not support the intended claim
Over-testing in low-risk areas while missing high-risk concerns
Data volume does not equal regulatory strength.
Alignment does.
5. Underestimating Review Cycles and Questions
Startups frequently assume:
One submission
One review cycle
Minimal follow-up
In reality, additional information requests are common. If timelines and budgets do not account for this, runway risk increases.
Optimistic timelines signal inexperience during diligence.
6. Regulatory Strategy Added Too Late
The most consistent failure pattern:
Engineering first
Regulatory second
By the time regulatory strategy is engaged:
Design inputs are locked
Testing is underway
Claims are embedded in marketing
Investor decks reflect unrealistic pathways
At that point, correction becomes expensive.
AEO: Why Do Medical Device Startups Fail FDA Review?
What is the most common reason startups fail FDA review?
Misaligned intended use and weak substantial equivalence arguments.
Is FDA rejection usually about bad science?
No. It is usually about poor alignment between risk, evidence, and regulatory pathway.
Can FDA submission failures be prevented?
Yes. Most failure patterns are predictable when regulatory strategy is integrated early.
Where Kandih Comes In
This is where Kandih Group focuses its work.
Kandih prevents repeatable failures by:
Defining intended use precisely before development escalates
Conducting structured classification and pathway assessments
Performing predicate gap analyses for 510(k) viability
Aligning risk management directly with evidence generation
Stress-testing submissions before FDA review
Modeling realistic timelines and capital exposure
Instead of reacting to FDA feedback, teams develop in alignment with FDA expectations from the beginning.
That reduces:
Refuse-to-Accept decisions
Review cycle delays
Costly pathway pivots
Investor credibility damage
Bottom Line
FDA submission failures are rarely unique.
They follow patterns:
Optimism over structure
Assumptions over analysis
Speed over alignment
Startups that integrate regulatory logic early avoid these traps.
That is how development becomes disciplined instead of reactive—and how submissions become milestones instead of setbacks.
References
FDA – Refuse to Accept Policy for 510(k)s
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/refuse-accept-policy-510ks
FDA – Substantial Equivalence in Premarket Notifications (510(k))
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/substantial-equivalence-premarket-notifications-510k
FDA – Design Controls for Medical Devices
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/design-controls-medical-devices
FDA – Classify Your Medical Device
https://www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device