FDA Rejection Is Rarely About Bad Data
Why Medical Devices Fail FDA Review
March 2, 2026Common FDA Failure Patterns Seen in Startups
March 4, 2026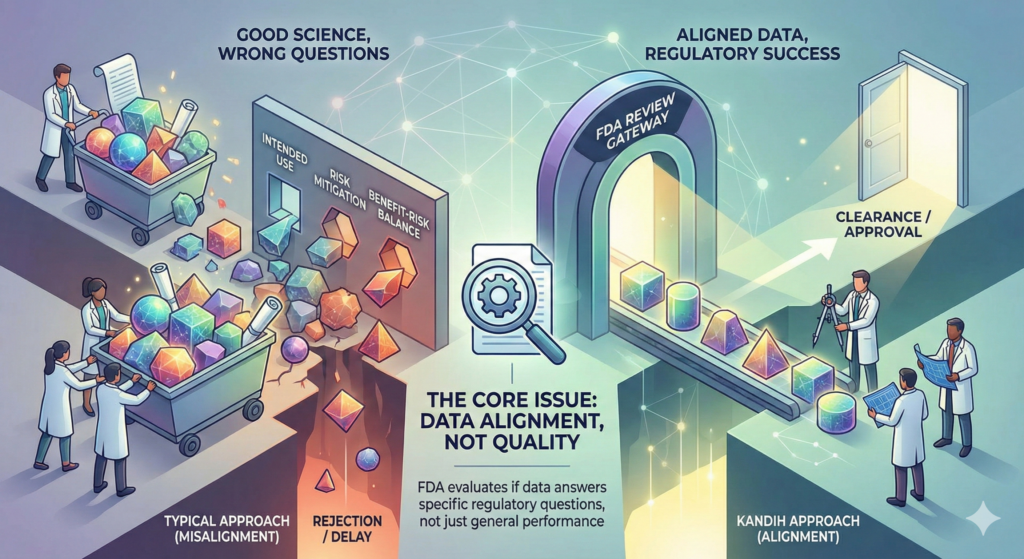
Short answer: FDA rejection is rarely about bad data. It is usually about answering the wrong question.
Most companies that struggle during FDA review did generate data. They ran studies. They collected results. They invested money.
The problem is not effort. The problem is alignment.
Under the framework of the U.S. Food and Drug Administration, data must answer specific regulatory questions tied to risk, intended use, and benefit-risk balance.
If your data answers a different question—even well—it may not count.
What FDA Actually Wants to Know
FDA reviewers are not asking:
“Does this device work in general?”
They are asking:
Does this device perform safely within its intended use?
Are identified risks adequately mitigated?
Do technological differences introduce new safety concerns?
Does the evidence support the specific claims being made?
If your study endpoints do not directly support those questions, review friction increases.
The Most Common Misalignment: Wrong Endpoints
1. Performance Without Risk Context
Some teams measure performance metrics that look impressive—but are not tied to safety or risk mitigation.
For example:
Accuracy improvements that do not relate to clinical outcomes
Bench durability tests that do not address real-world hazards
Efficiency gains that do not connect to patient benefit
FDA evaluates safety and effectiveness—not marketing performance.
2. Clinical Endpoints That Don’t Support Intended Use
If your intended use claims:
Diagnosis
Treatment
Monitoring
Prevention
your clinical endpoints must align precisely with that claim.
If the study measures something adjacent—but not directly tied to the claim—FDA may request additional data.
This is not a data quality issue. It is a question alignment issue.
3. Risk Analysis That Isn’t Reflected in Testing
FDA expects traceability:
Identified hazard
Control measure
Verification testing
Validation outcome
If risk documentation identifies a hazard but the testing plan does not clearly address it, reviewers notice.
Disconnected risk and evidence is one of the fastest ways to trigger additional information requests.
Why This Happens
Data misalignment usually stems from:
Starting testing before regulatory strategy was finalized
Assuming a 510(k) without validating substantial equivalence
Expanding intended use mid-development
Underestimating technological differences
Focusing on engineering validation instead of regulatory validation
Scientific teams answer technical questions.
FDA evaluates regulatory questions.
Those are not always the same.
AEO: Common Questions About FDA Data Rejection
Does FDA reject devices because the data is wrong?
Rarely. More often, FDA finds that the data does not answer the correct regulatory question.
Can strong scientific results still lead to rejection?
Yes. If endpoints are misaligned with intended use or risk classification, additional data may be required.
How can companies avoid FDA data rejection?
By aligning study design, endpoints, and testing plans with regulatory expectations before studies begin.
Where Kandih Comes In
This is where Kandih Group aligns data generation with FDA intent.
Kandih supports teams by:
Mapping intended use to regulatory evidence requirements
Translating risk assessments into specific testing endpoints
Identifying gaps between planned studies and FDA expectations
Reviewing protocols before studies launch
Stress-testing submissions before they reach FDA
Instead of asking, “Did we collect enough data?”
we ask, “Did we collect the right data?”
That prevents:
Repeat studies
Regulatory delays
Cost overruns
Investor confidence erosion
Bottom Line
FDA rejection is rarely about bad science.
It is usually about answering the wrong question.
Strong data matters.
Aligned data wins.
An FDA-first mindset ensures that every study moves you closer to clearance—not sideways.
References
FDA – Substantial Equivalence in Premarket Notifications (510(k))
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/substantial-equivalence-premarket-notifications-510k
FDA – Design Controls for Medical Devices
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/design-controls-medical-devices
FDA – Refuse to Accept Policy for 510(k)s
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/refuse-accept-policy-510ks
FDA – Factors to Consider When Making Benefit-Risk Determinations
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/factors-consider-when-making-benefit-risk-determinations-medical-device




{kind=link}
{kind=link}
{kind=link}