The Questions Nobody Is Asking at the Whiteboard
Environmental Risk Assessment in Drug Development: What It Is, Why It Matters, and When to Start
April 20, 2026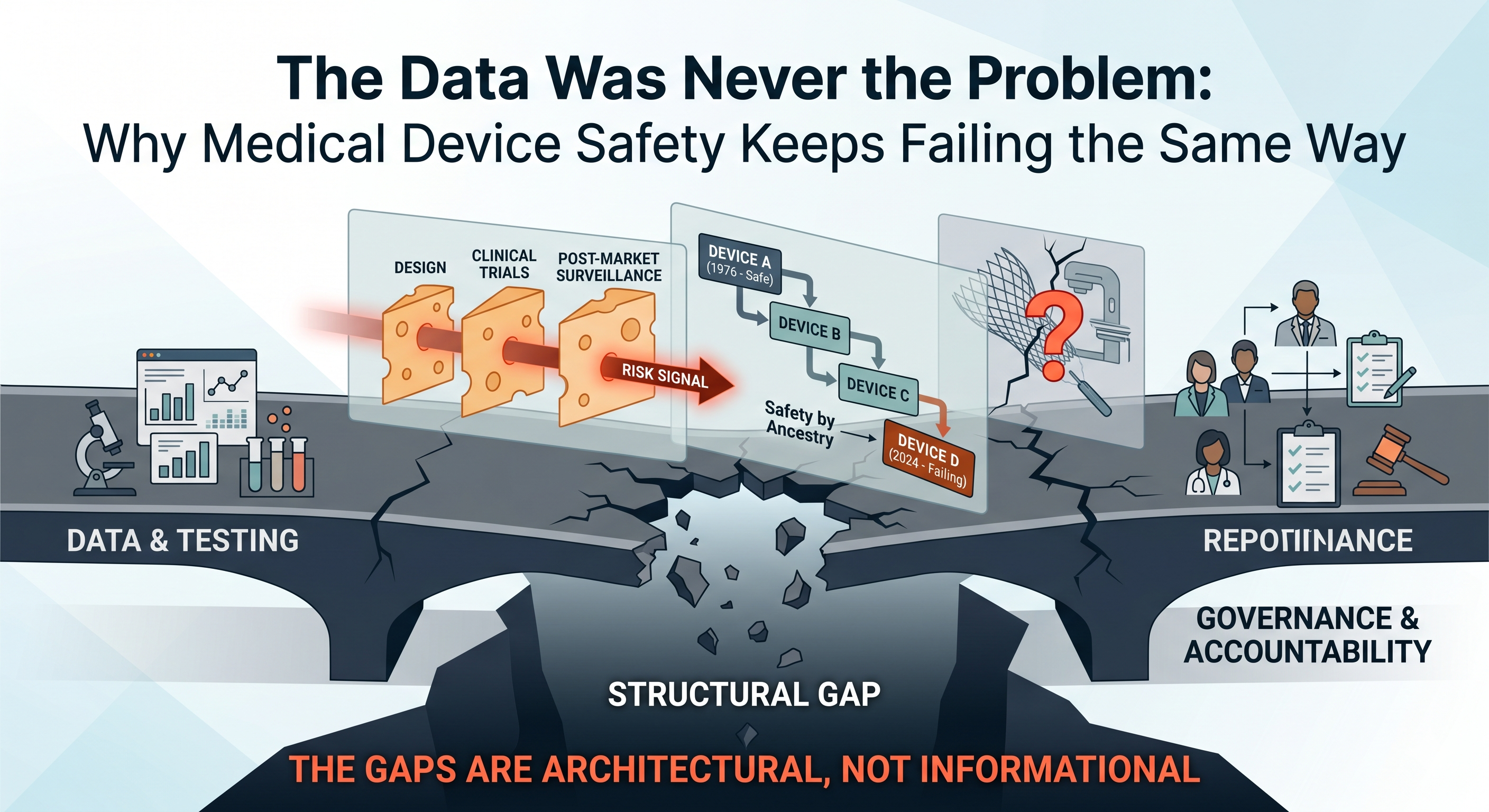
The Data Was Never the Problem: Why Medical Device Safety Keeps Failing the Same Way
April 22, 2026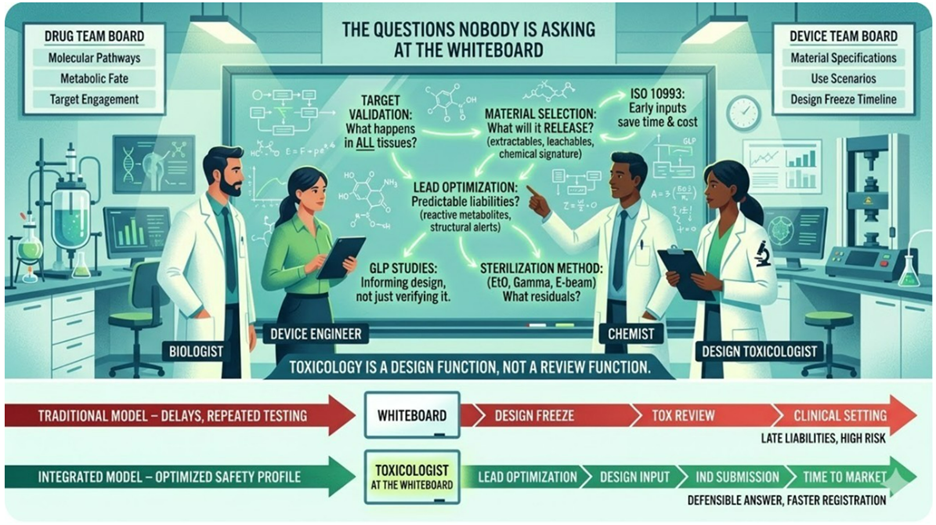
I design FDA evidence that protects capital · Regulatory Toxicologist, PhD DABT · Medical Devices · Diagnostics · Drugs · Biologics · Medical Foods · Founder & CEO, Kandih BioScience
There is a room where medical product development actually begins. It is not the GLP facility. It is not the pre-IND meeting or the Q-Submission. It is not the CRO’s project kickoff call or the design verification review.
It is a conference room, early in the life of a program, where a small team stands in front of a whiteboard. For a device team, it is covered in materials specifications, use scenarios, and a design freeze timeline. For a drug/biologic team, the board is covered in molecular structures and pathway diagrams. The energy is the same. So is the problem.
Nobody in the room is a toxicologist.
That is not unusual. It is standard. And it is the reason so many programs that looked scientifically sound at the whiteboard arrive at regulatory submission — or worse, in a clinical setting — carrying liabilities that were never invisible. They were just never named.
What toxicology actually is at the design stage
Ask most founders when they think about engaging toxicology and they will describe a checkpoint. For device developers, it is the biocompatibility testing battery required for a 510(k) or PMA submission. For drug developers, it is the IND-enabling studies. Something you commission, complete, and file. That framing is not wrong. It is just about two to three years too late.
Toxicology at the design stage is not a review function. It is a design function.
For devices, the decisions made during early design — which materials to specify, which sterilization method to use, how long the device will contact tissue and which tissues — these decisions are your biocompatibility evaluation.
For drugs/biologic, the decisions made at lead optimization — which scaffold to advance, which metabolic pathway you are committing to, which species will inform your human safety prediction — these decisions are your toxicity profile.
They are not inputs to a future testing program. They are the program, written in advance, before a single study is run.
By the time a toxicologist reviews your submission package, those decisions are fixed. The question at that stage is not what should we build? It is can we defend what we built? Those are different questions.
The first is strategic. The second is damage control.
What the whiteboard is missing
There are questions that belong at the beginning of a program — for all medical products including devices — that almost never get asked there.
For device programs: what will this material release into the body, under what conditions, and for how long? The extractables and leachable profile of a device is not determined at the testing stage. It is determined at material selection — when nobody thinks to ask a toxicologist because the conversation feels like engineering, not safety.
For drug programs: what does your target do in every tissue where it is expressed, not just the tissue you are trying to treat? What does your lead structure tell you about its metabolic fate before you run a single assay? Certain structural features are associated with known liabilities — reactive metabolite formation, covalent binding to proteins — that are readable in the chemistry, by someone trained to read them, at the whiteboard stage.
Sterilization method is another decision that feels operational until it isn’t. Ethylene oxide, gamma radiation, e-beam — each leaves a different chemical signature on device materials. EtO residuals are a regulatory endpoint under ISO 10993-7. The choice of sterilization method is made early for manufacturing reasons; it determines whether that endpoint exists in your submission package and what it will say. That is a toxicology-informed decision. It rarely gets treated as one.
These are not regulatory questions. They are scientific questions with regulatory consequences. And they have a window — a narrow one — in which answering them changes the program rather than just documenting its history.
What Rezulin’s chemistry already knew
In 2000, troglitazone — sold as Rezulin — was withdrawn from the US market after causing severe drug-induced liver injury, including deaths. It had been approved three years earlier for type 2 diabetes and taken by tens of thousands of patients.
What makes this case worth sitting with is not the withdrawal. It is what happened within the same drug class. Rosiglitazone and pioglitazone — developed targeting the same receptor — did not produce the same hepatotoxic signal.
Same mechanism of action. Different structural decisions at lead optimization. Different outcome.
The structural features that distinguished troglitazone were associated with metabolic activation pathways that generated reactive metabolites capable of binding to liver proteins. These were not unknowable. In the years following the withdrawal, the pharmaceutical industry systematically implemented early-stage screening for reactive metabolites — at exactly the stage where the question should have been asked the first time.
The corrective action was taken at the stage where the original question belonged.
Device development has its own version of this story. Materials that perform well mechanically and pass initial cytotoxicity screening have later been found to generate degradation products or leachable under real-use conditions — extended implantation, repeated sterilization cycles, patient-specific biological environments — that were not characterized early because nobody mapped the full exposure scenario at the design stage.
The signals were in the chemistry. They were just not read at the whiteboard.
What changes when the right person is in the room
A toxicologist at the design stage does not slow down development. They redirect it.
For device programs: they identify which ISO 10993 endpoints apply to this specific device — based on contact type, duration, and tissue — before materials are finalized, not after design freeze. They flag that a sterilization choice will generate a residuals endpoint. They identify whether existing material data can support a test waiver or whether new testing is required — a question with significant cost and timeline implications that is almost never asked until a submission is being assembled.
For drug programs: they flag structural alerts before significant synthesis effort is committed to a scaffold carrying a known liability. They inform lead optimization criteria so that reactive metabolite potential is a selection filter, not a post-hoc finding.
Most importantly, for both drugs and devices, they think forward from the whiteboard to the regulatory package — asking not just whether the product is safe enough to advance, but whether the data being generated will be capable of producing a defensible answer to that question.
That is architectural thinking. It cannot be retrofitted after design freeze. It cannot be retrofitted after candidate selection. The question worth adding to your next whiteboard session. Not when do we need to file?
What does toxicology need to know before we commit to this design?
The first question treats safety as a destination on the development timeline. The second treats it as a design input — one that shapes what gets built, not just whether it clears regulatory review.
The whiteboard is where medical product development begins. It is also where safety is either built into the architecture or left for someone else to find later — in a very different kind of room, under very different circumstances. That conversation is not too early to have at the whiteboard. It is the only moment early enough to have it.
#RegulatoryToxicology #DrugDevelopment #MedicalDevices #Biotech #Pharma #RegulatoryStrategy #PreclinicalDevelopment #ISO10993 #IND #510k #SafetyByDesign #KandihGroupIf your team is approaching design freeze or candidate selection without toxicology input, that window is closing. Happy to discuss what early engagement looks like in practice




{kind=link}
{kind=link}
{kind=link}