The Data Was Never the Problem: Why Medical Device Safety Keeps Failing the Same Way

The Questions Nobody Is Asking at the Whiteboard
April 21, 2026
Safety is Not Advice: Why Better Data Won’t Save Your Drug Without Diagnostic Infrastructure
April 23, 2026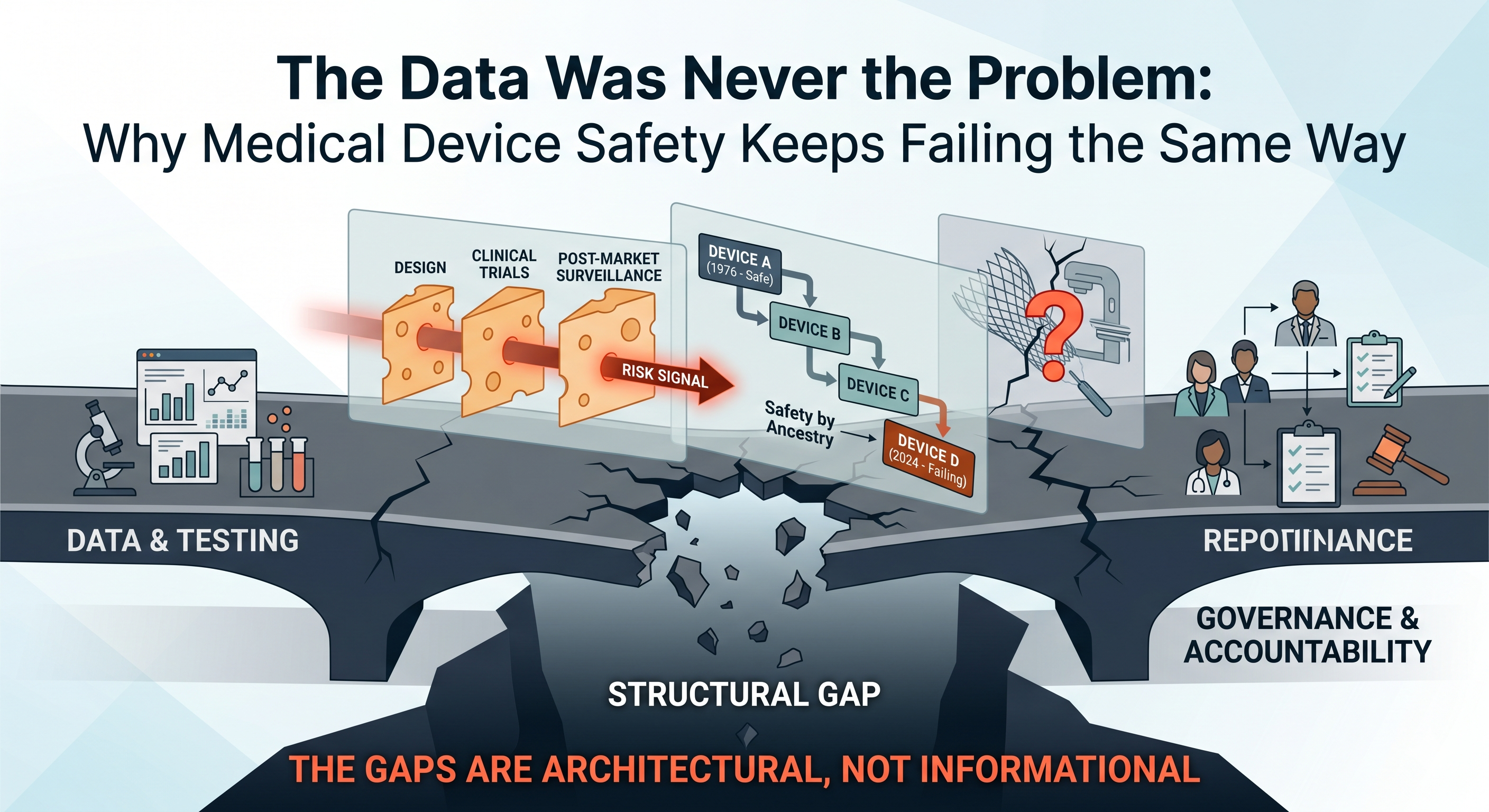
Why “more testing” won’t save your device program without diagnostic infrastructure and accountability
Every major medical device safety disaster has a predictable anatomy — and it’s the same anatomy as drug safety disasters. From the Dalkon Shield in the 1970s to metal-on-metal hips in the 2000s, from Therac-25’s lethal radiation overdoses to Philips’ contaminated CPAP machines, the pattern repeats: warning signs existed, the science was available, and no one with authority was accountable to act on it. The device world just has its own particularly insidious ways of letting danger through.
If you’re building or investing in a medical device company, you need to understand something uncomfortable: “we tested it” is not the same as “we built a system to catch what could go wrong.” The device regulatory landscape has structural features — the 510(k) predicate chain, the fragmentation of post-market surveillance, the speed of hardware iteration — that make accountability failures not just possible, but almost architecturally inevitable unless you deliberately engineer against them.
This isn’t about bashing regulators or manufacturers. It’s about recognizing that the system, as currently constructed, treats safety as a checkpoint rather than a continuous governance function. And checkpoints can be passed by products that later hurt people — not because anyone was malicious, but because no one was structurally accountable for the risks that fell between the cracks.
“If It’s Dangerous, Testing Will Catch It” — The Device Version of a Costly Myth
Many device founders assume that if their product is dangerous, the combination of bench testing, biocompatibility studies, and clinical data will surface the problem before it reaches patients. This assumption rests on the same flawed logic that haunts drug development: the belief that generating data is the same as building the infrastructure to act on what the data reveals.
In medical devices, the diagnostic infrastructure for safety spans three layers — each with its own characteristic failure modes:
Design-Phase Predictivity. Are your bench tests, simulations, and biocompatibility assays actually capable of detecting the plausible failure modes of your specific device, in the specific anatomical and physiological context where it will be used? Or are you running a generic battery of tests that might miss a mechanism-specific risk?
Clinical Visibility. If your device does cause harm, are your clinical studies designed to see it? Are endpoints defined to capture not just efficacy but also the specific types of adverse events your device’s mechanism could plausibly produce? Are you studying the device in the patient populations most likely to reveal problems — or only in the healthiest, lowest-risk subjects?
Post-Market Detection. Once the device is in widespread use, do you have systems in place to detect safety signals before they become mass casualties? Or are you relying on passive adverse event reporting — the same system that repeatedly fails to catch problems until thousands of patients are affected?
If any of these layers is weak, safety becomes a post-mortem exercise — understanding what went wrong only after patients are hurt. History provides stark examples of how gaps in each layer have led to disaster, and unlike drug safety failures, device failures often involve physical objects permanently implanted in human bodies — objects that can’t be “discontinued” without additional surgery.
The Dalkon Shield: No Infrastructure, No Accountability, No Protection
Before there was a modern device regulatory framework in the United States, the Dalkon Shield happened — and it’s worth understanding why, because the regulatory architecture that exists today was built in direct response to this disaster. And even that architecture, as we’ll see, has proven insufficient.
The Dalkon Shield was an intrauterine contraceptive device (IUD) developed in the late 1960s by physician Hugh Davis and engineer Irwin Lerner, and later marketed aggressively by the A.H. Robins Company. Its design featured a distinctive crab-claw shape intended to reduce expulsion rates, and a multifilament tail string — a bundle of fine fibers encased in a nylon sheath — that allowed for removal.
The problem was that this multifilament design acted as a wick. Previous IUD designs had used monofilament strings precisely to prevent bacteria from traveling from the non-sterile vaginal canal into the sterile uterine environment. The Dalkon Shield’s tail, by contrast, created a highway for pathogens. The result: dramatically elevated rates of pelvic inflammatory disease (PID), septic abortions, infertility, and in some cases, death.
What makes the Dalkon Shield a diagnostic infrastructure failure rather than merely a “bad product” story is how every layer of the system failed simultaneously.
At the design phase, the tail string’s wicking risk was knowable — and arguably known. The biocompatibility and mechanical behavior of multifilament versus monofilament strings in a warm, moist, bacteria-rich environment was not exotic science. But no independent testing authority existed to evaluate the design before it went to market. At the time, the FDA had authority over drugs but not devices. The Dalkon Shield reached millions of women without a single requirement for rigorous safety testing.
At the clinical evidence phase, Davis’s own published study — which reported an impressively low 1.1% pregnancy rate — was riddled with methodological problems. The study enrolled too few participants for statistical significance, the average enrollment period was five months of a twelve-month study, women who dropped out weren’t counted, and Davis had recommended spermicide use during peak fertility — artificially deflating the pregnancy rate. Most critically, Davis did not disclose his financial interest in the device in his published paper. The study was not peer-reviewed before publication. This wasn’t a case of honest scientific uncertainty; it was promotional material masquerading as evidence.
At the post-market level, there was essentially no surveillance system. Physicians across the country began reporting devastating complications — traumatic insertions, uterine perforations, severe infections — but there was no centralized mechanism to aggregate these reports, detect the pattern, or trigger action. A.H. Robins suspended domestic distribution in June 1974 at the FDA’s request, but there was no formal recall until 1980 — six years during which devices already implanted continued to cause harm, and international distribution continued.
The Dalkon Shield ultimately affected an estimated 2.2 million women in the United States alone, generated roughly 327,000 injury claims, and forced A.H. Robins into bankruptcy. More importantly for our purposes, it directly catalyzed the Medical Device Amendments of 1976, which gave the FDA authority to regulate medical devices and established the classification system (Class I, II, III) and clearance pathways (including the 510(k)) that govern devices today.
The lesson should have been definitive: without diagnostic infrastructure — without the systems that force safety evidence to be generated, evaluated, and acted upon — devices will reach patients based on marketing claims rather than safety data. The 1976 Amendments were supposed to fix this. What happened next shows why legislative frameworks alone aren’t enough.
The 510(k) Predicate Trap: Safety by Ancestry
The single most structurally dangerous feature of the modern device regulatory landscape is the assumption that substantial equivalence equals substantial safety.
The FDA’s 510(k) pathway — which clears roughly 90% of moderate-risk devices that reach the U.S. market — allows a new device to bypass full clinical testing (the more rigorous Premarket Approval, or PMA, pathway) if the manufacturer can demonstrate that it is “substantially equivalent” to a device already legally marketed. The logic sounds reasonable: if the predicate device was safe, and yours is materially similar, the risk profile should be comparable.
The problem is that predicates can chain. Device B references Device A as its predicate. Device C references Device B. Device D references Device C. By the time you reach Device D, it may bear little functional resemblance to Device A — but it has inherited A’s presumption of safety without anyone ever conducting a definitive safety evaluation on the product that patients actually receive. This is not a theoretical concern. It is the mechanism by which entire device categories have drifted into danger.
Compounding the structural problem is a practical one: the 510(k) pathway was designed for an era of incremental mechanical innovation. It assumes that if the materials are similar and the intended use is similar, the risks will be similar. But this assumption breaks down when a device is moved to a new anatomical site, used in a different patient population, or when the cumulative effect of many small design changes alters the risk profile in ways that no single change would have triggered a new safety review.
This is exactly what happened with transvaginal pelvic mesh — and the consequences were catastrophic.
Pelvic Mesh: The Predicate Chain That Broke
Surgical mesh for pelvic organ prolapse (POP) and stress urinary incontinence (SUI) was cleared through the 510(k) pathway by referencing predicate devices originally designed for abdominal hernia repair. The regulatory logic: mesh is mesh, the material is biocompatible, and it has a well-established safety record in the abdominal wall. No one was required to run clinical trials demonstrating that mesh engineered for abdominal fascia would perform safely when implanted against the delicate, hormonally responsive, mechanically distinct tissue of the vaginal wall.
The science that would have predicted problems was not obscure. Bioengineers and urogynecologists understood that pelvic tissue differs fundamentally from abdominal wall tissue. The mechanical stresses are different — the pelvic floor is dynamic, load-bearing, and subject to forces from standing, walking, coughing, and sexual activity. The tissue composition is different — vaginal mucosa is thinner, more vascular, and hormonally responsive, particularly in the postmenopausal women who are the primary candidates for prolapse repair. The inflammatory and healing responses differ. The proximity to critical structures — bladder, urethra, bowel, nerves — creates failure consequences that don’t exist in abdominal hernia repair.
None of this mattered under the 510(k) framework because no one was required to ask these questions, let alone answer them. The predicate chain carried the products to market on an assumption — substantial equivalence — that no one was accountable to verify.
The result was catastrophic. Tens of thousands of women experienced chronic pain, mesh erosion through vaginal tissue, infections, hemorrhage, dyspareunia (painful intercourse), urinary problems, and bowel complications — injuries so severe that many required multiple corrective surgeries, and some were left with permanent disability. The adverse event reports accumulated for years. Individual clinicians and patient advocates raised alarms. Peer-reviewed literature documented the complication rates. But the regulatory response was slow.
The FDA issued a Public Health Notification in 2008 — noting over 1,000 adverse event reports related to mesh erosion through vaginal tissue in the prior three years — but did not remove the products. In 2011, the FDA issued a Safety Communication stating that serious complications were “not rare” and that it was “not clear” that mesh repairs were more effective than traditional surgery for POP. It took until 2016 for the FDA to reclassify pelvic mesh for POP from Class II (510(k) clearance) to Class III (requiring PMA with clinical data). And it wasn’t until April 2019 — more than a decade after the signals were unmistakable — that the FDA ordered the remaining manufacturers to stop distributing pelvic mesh for transvaginal POP repair.
The mesh debacle generated hundreds of thousands of lawsuits and billions of dollars in settlements, but the cost in human suffering — chronic pain, loss of sexual function, repeat surgeries, psychological trauma — is incalculable. For our purposes, the critical takeaway is structural: the 510(k) predicate system, by design, allowed a product to reach a fundamentally different anatomical environment without any requirement that safety be independently demonstrated in that environment. The diagnostic infrastructure wasn’t weak; it was absent. And the accountability infrastructure — who has the authority and obligation to question a predicate chain — was undefined.
Therac-25: When Software Becomes the Safety System — and Fails
If the Dalkon Shield represents the failure of a pre-regulatory era, and pelvic mesh represents the failure of a regulatory shortcut, the Therac-25 represents a different and increasingly urgent category: what happens when software replaces hardware safety systems and no one is accountable for validating that substitution.
The Therac-25 was a computer-controlled linear accelerator for radiation therapy, developed by Atomic Energy of Canada Limited (AECL) and deployed in the United States and Canada beginning in 1982. It was the most advanced radiation therapy machine of its era — faster, more versatile, and more computerized than its predecessors, the Therac-6 and Therac-20. Between 1985 and 1987, it massively overdosed at least six patients, delivering radiation doses hundreds of times greater than prescribed. At least four patients died. Others suffered severe radiation burns, tissue necrosis, and lifelong disability.
The technical cause was software bugs — specifically, race conditions in the machine’s control software that could, under certain operator input sequences, result in the high-energy electron beam being activated without the beam-spreading or beam-attenuating hardware in the correct position. When this happened, instead of receiving a diffuse therapeutic dose, the patient was hit with a concentrated, needle-thin beam of radiation at full power.
But the deeper cause was an accountability failure that echoes across every case in this article.
The Therac-25’s predecessors — the Therac-6 and Therac-20 — had relied on independent hardware interlocks as the primary safety mechanism. Physical switches, fuses, and mechanical positioning guides ensured that the beam could not activate if the treatment head was in the wrong configuration. These hardware safeguards worked. The software was a convenience layer, not a safety-critical system.
With the Therac-25, AECL made a fateful architectural decision: the hardware interlocks were removed, and safety was delegated entirely to software. The rationale was that the software could handle safety checks faster and more flexibly than mechanical systems. But the software had never been independently reviewed or rigorously tested. It was developed by a single programmer using PDP-11 assembly language, evolving from earlier codebases over several years. There was no formal software safety analysis. No independent verification. No separation between the software that controlled treatment delivery and the software that was supposed to prevent unsafe states.
When patients reported being “burned” by the machine, AECL’s initial response was denial. The company sent a letter to one hospital stating that radiation overdose was “impossible” — citing the very software that was causing the overdoses. When operators described the malfunction pattern, AECL engineers couldn’t reproduce it — because the bug was a race condition triggered by a specific timing of keystrokes that was vanishingly unlikely but not impossible. AECL made incremental fixes after each reported incident but failed to conduct a systematic review of the software architecture.
The FDA’s role was constrained by its own limitations. At the time, the agency had limited expertise in software safety and limited authority to mandate software design reviews. The Therac-25 had been cleared partly through a substantial-equivalence argument to its predecessors — the same predecessors whose hardware safety systems had been removed. The 510(k) framework, once again, allowed a fundamental safety architecture change to pass through without dedicated evaluation.
It took six overdoses, multiple deaths, and an investigation by MIT professor Nancy Leveson — whose subsequent analysis became a foundational text in software safety engineering — before the machines were finally shut down in February 1987 and subjected to a comprehensive corrective action plan including, critically, the reinstallation of independent hardware safety interlocks.
The Therac-25 case established several principles that remain urgently relevant as software-driven and AI-enabled medical devices proliferate:
Software cannot be its own safety system. When the same software that delivers treatment is also responsible for preventing unsafe treatment, a single failure mode can be catastrophic. Independent, redundant safety layers — whether hardware interlocks, separate monitoring software, or human-in-the-loop checks — are essential.
“No reported failures” does not mean “safe.” AECL’s confidence was built on the absence of evidence rather than evidence of absence. The race condition that caused the overdoses was extremely difficult to reproduce in testing — but it was not impossible in clinical use. For software-controlled devices, exhaustive testing cannot prove safety; only rigorous architectural design, formal verification, and defense-in-depth can provide adequate assurance.
The manufacturer’s culture is part of the safety system. AECL’s repeated insistence that overdoses were “impossible” — even after patients and operators reported them — delayed corrective action by months. When the organization responsible for safety doesn’t believe its product can fail, it will dismiss or explain away every signal until the evidence is overwhelming.
Metal-on-Metal Hips: The Signal Was There — The System Looked Away
If the Therac-25 is the canonical software failure, the DePuy ASR hip replacement system is the canonical accountability failure — the device equivalent of Vioxx. It’s the case that demonstrates most clearly what happens when signals exist, are visible, and are systematically deprioritized in favor of commercial momentum.
Metal-on-metal (MoM) hip implants were marketed as a superior alternative to traditional metal-on-polyethylene designs, particularly for younger, more active patients. The pitch was compelling: metal bearing surfaces were harder and more wear-resistant than polyethylene, potentially lasting longer and allowing for larger femoral head sizes that would improve range of motion and reduce dislocation risk.
But the mechanism of potential harm was foreseeable from basic materials science. Two metal surfaces — typically cobalt-chromium alloys — grinding against each other under the cyclical loads of human movement generate metallic debris: cobalt and chromium particles and ions that can infiltrate surrounding soft tissue. This was known. The biological consequences of chronic exposure to cobalt and chromium ions — metallosis, adverse local tissue reactions (ALTRs), pseudotumors, osteolysis, and systemic toxicity — were described in the orthopedic literature before the ASR was ever implanted.
DePuy (a Johnson & Johnson subsidiary) launched the ASR hip resurfacing system in 2003 and the ASR XL total hip replacement in 2005. From early in the product’s life, the signals were visible:
Internal data showed problems. DePuy’s own post-market data showed higher-than-expected revision rates. Internal documents later revealed in litigation indicated that the company was aware of design concerns, including the shallow acetabular cup geometry that increased edge loading and metal debris generation.
Registries flagged the risk. National joint registries — particularly the Australian Orthopaedic Association National Joint Replacement Registry and the UK’s National Joint Registry — published data showing that the ASR had significantly higher revision rates than competing implants. The Australian registry data was publicly available and showed the ASR performing worse than virtually every other hip system on the market.
Clinicians reported adverse events. Orthopedic surgeons reported unusual tissue reactions, unexplained pain, elevated blood cobalt and chromium levels, and pseudotumor formation in ASR patients. These reports were not centralized or systematically aggregated — they were scattered across individual adverse event reports, conference presentations, and case series.
The scientific literature raised concerns. Published studies documented the mechanisms of metal debris generation, the biological response to cobalt-chromium particles, and the specific design features (particularly small cup sizes and steep cup angles) that exacerbated the problem.
Despite all of this, the ASR remained on the market until August 2010, when DePuy issued a voluntary recall — not in response to a regulatory order, but under mounting legal and reputational pressure. By that time, approximately 93,000 ASR devices had been implanted worldwide. The recall triggered one of the largest medical device mass tort litigations in history, ultimately costing Johnson & Johnson billions of dollars in settlements.
The core failure was not informational. The information existed. The failure was structural: no one in the system had both the clear sight of the accumulating evidence and the unambiguous authority — and obligation — to stop the commercial distribution of a profitable product.
DePuy’s internal teams saw the revision rate data but were operating within an organization whose dominant incentive was commercial success. The FDA’s post-market surveillance relied heavily on the Manufacturer and User Facility Device Experience (MAUDE) database — a passive, voluntary reporting system with well-known limitations including underreporting, inconsistent data quality, and no denominator (you know how many reports came in, but not how many devices are in use, making rate calculations impossible). National registries had the data but no enforcement authority. Individual surgeons had clinical observations but no mechanism to aggregate them into actionable signals.
The diagnostic infrastructure existed in fragments — registries here, adverse event reports there, published case series elsewhere — but no governance system connected the dots and compelled action. Safety insights were available but functioned as advice, not as orders. And advice, as the Vioxx saga demonstrated in the drug world, is easily overridden by commercial momentum and organizational optimism.
Philips CPAP: A Modern Disaster, a Familiar Pattern
Lest anyone think these accountability failures are relics of earlier, less sophisticated eras, the Philips Respironics CPAP recall — unfolding from 2021 to the present — demonstrates that the pattern is alive, well, and affecting millions of patients right now.
In June 2021, Philips Respironics issued a voluntary recall of millions of CPAP, BiPAP, and mechanical ventilator devices after determining that the polyester-based polyurethane (PE-PUR) foam used for sound dampening could degrade over time, particularly in conditions of heat and humidity. Degraded foam could release small black particles into the device’s air pathway — particles that patients would inhale directly into their lungs night after night — as well as volatile organic compounds (VOCs) with potential toxic and carcinogenic effects.
The scale was staggering: approximately 15 million devices worldwide were affected, including some of the most widely prescribed sleep apnea machines on the market. The FDA has received over 116,000 reports of problems, including more than 560 reports of deaths potentially associated with the foam degradation.
But the most damning aspect of the Philips case isn’t the scale — it’s the timeline of known risk.
An FDA inspection of Philips’ Pennsylvania offices revealed that the company had been aware of the foam degradation issue years before the recall. As early as 2015, Philips received customer warnings about foam degradation in its Trilogy ventilator products. Between 2016 and early 2021, the FDA found fourteen separate instances in which Philips was made aware of the issue or was analyzing it internally. In each case, no further design changes, corrective actions, or field corrections were implemented.
Perhaps most damningly, Philips’ own foam supplier — William T. Burnett — wrote to Philips in a 2018 email stating plainly that they would not recommend use of polyester foam in such an environment and that it would eventually decompose. The supplier told them. And still, nothing changed until 2021.
This is the ranitidine pattern applied to devices. A material-level risk, knowable through basic chemistry and explicitly flagged by a supplier, persisted for years because the organizational infrastructure did not compel action. The people who knew about the risk did not have — or did not exercise — the authority to force a design change, a field action, or even a formal risk assessment. Complaints were handled “on a case-by-case basis,” which is the organizational equivalent of hoping the problem will go away if you address each instance individually and never look at the aggregate.
When Philips finally acted, the remediation was itself compromised. The company chose to replace the PE-PUR foam with a silicone-based foam — but an FDA inspection discovered that a device with the new foam had failed emissions testing, producing compounds of concern with carcinogenic properties. Philips had not disclosed this test result to the FDA. The pattern of information asymmetry — the company knowing more about the risks than the regulator or the patient — persisted even during the corrective action.
The Philips case is not about a rare, unpredictable failure. It’s about a common, predictable material degradation that was flagged repeatedly, by multiple sources, over multiple years, and was not acted upon because no governance system demanded action. The diagnostic infrastructure (analytical chemistry, supplier expertise, complaint trending) could have caught this at any point. The accountability infrastructure — who reviews material stability data, who triggers a risk assessment when a supplier says “don’t use this material,” who decides when a pattern of complaints becomes a signal — was either absent or non-functional.
The Core Problem: Testing vs. Governance
These cases — spanning five decades, multiple device categories, and every failure mode from material degradation to software bugs to design-level anatomical mismatch — share a single root cause: the people who saw the risk didn’t have the power (or the mandate) to act on it, and the people who had the power didn’t see the risk (or didn’t want to).
This is not a data problem. In almost every case, the relevant data existed or could have been generated with modest effort. It’s a governance problem — a failure to build systems that connect safety information to decision authority with enforceable accountability.
In the device world, this governance gap is amplified by several structural factors that don’t have exact parallels in drug development:
The 510(k) pathway incentivizes minimal safety evidence. When you can reach market by demonstrating substantial equivalence rather than independent safety, the organizational incentive is to generate the minimum evidence needed for clearance — not the maximum evidence needed to understand your product’s risk profile. Companies that voluntarily generate more safety data than required are effectively penalizing themselves with higher costs and longer timelines relative to competitors who don’t.
Post-market surveillance is fragmented and passive. The MAUDE database — the FDA’s primary post-market device surveillance system — relies on voluntary reporting by manufacturers, healthcare facilities, and patients. It has no denominator data, making rate calculations impossible. It suffers from massive underreporting. It lacks standardized terminology, making signal detection difficult. And it operates with significant time lags. By the time a pattern becomes visible in MAUDE, thousands of patients may already be affected.
Devices are physical objects that can’t be “discontinued” like drugs. When a drug is withdrawn, patients stop taking it. When an implantable device is recalled, the device is still inside the patient. Explantation surgery carries its own risks — sometimes greater than the risks of leaving the device in place. This creates a perverse dynamic where the worse the potential harm, the harder it is to take corrective action, because the corrective action itself is harmful. The Björk-Shiley heart valve illustrated this dilemma starkly: a mechanical heart valve with a known risk of catastrophic strut fracture, where removing the valve could itself be fatal. Patients lived for decades knowing they had a potential “time bomb” in their chests because the alternative — open-heart surgery to replace the valve — was itself life-threatening.
Software and AI introduce failure modes that traditional device testing doesn’t capture. The Therac-25 demonstrated in the 1980s that software bugs in medical devices can be lethal. Today, as AI/ML-enabled devices proliferate — diagnostic algorithms, treatment recommendation systems, autonomous surgical robots — the failure modes multiply. An AI model can degrade silently as the patient population shifts (dataset drift). A software update can introduce new failure modes in a device that was previously safe. The traditional device paradigm of “test the product, clear it, monitor it” assumes the product is static. Software is not static, and the regulatory framework is still catching up to this reality.
The Regulatory Toxicologist’s Device Equivalent: The Safety Systems Engineer
In drug development, the regulatory toxicologist should function as the governance engine for safety — the person who maps plausible harms, designs detection into studies, builds escalation criteria, and holds the authority to pause or redesign a program when signals emerge.
The medical device equivalent is less well-defined organizationally, which is itself part of the problem. But the function is the same, and in a truly safety-centric device company, it should be embodied by a combination of roles — call it the safety systems engineering function — that collectively performs the following:
Map plausible failure modes comprehensively. Not just the failure modes that bench testing will catch, but the ones that arise from the interaction of the device with human anatomy, human behavior, clinical workflows, and time. What happens when tissue grows into the device? What happens when the device is used by an operator who’s fatigued, undertrained, or in a hurry? What happens when the device degrades after five years, ten years, twenty years? What happens when the device is used in a patient population different from the one studied in clinical trials — older, sicker, anatomically different?
Design detection into the entire product lifecycle. Bench testing is necessary but not sufficient. Clinical studies must include endpoints and monitoring protocols designed to catch the specific types of harm your device could plausibly cause — not just generic safety endpoints. Post-market surveillance must be active, not passive: registries, scheduled follow-up imaging, blood biomarkers (like cobalt and chromium levels for metal implants), patient-reported outcome measures. If you’re only going to learn about problems when patients or surgeons voluntarily report them to a database, you are going to learn about problems too late.
Build clear escalation criteria in advance. What revision rate triggers a design review? What adverse event frequency triggers a field action? What complaint trend triggers a formal risk assessment? If these thresholds aren’t defined before you launch, you will spend your post-market phase debating whether each signal is “real” rather than acting on it. Pre-defined stopping rules remove ambiguity and prevent the organizational tendency to move the goalposts when inconvenient data arrives.
Own the biocompatibility and materials story over time. Devices aren’t static chemistry experiments; they exist in a dynamic biological environment. Materials degrade. Corrosion products accumulate. Tissue responses evolve. The Philips CPAP case shows what happens when material stability is assumed rather than monitored. A robust safety function tracks the long-term behavior of every material in the device — not just at the point of clearance, but throughout the product’s market life.
Maintain independent authority to escalate. This is the most critical and most frequently absent element. Someone in the organization must have the explicit mandate and the executive support to say “stop” or “change course” when safety data warrants it — even when the product is commercially successful, even when the sales team is hitting targets, even when the board is counting on next quarter’s revenue. If safety authority is diffuse, the default trajectory is commercial: ship more, sell more, hope for the best. The DePuy ASR story is what happens when no one has the power to override that trajectory.
If your device company doesn’t have people performing these five functions with real authority, you don’t have a safety program. You have a hope-based compliance exercise.
Founders and Investors: Ask the Right Questions
The cases above aren’t curiosities for regulatory historians. They are templates for the next device safety disaster — which is already forming somewhere in the pipeline, in a company that believes its testing is adequate, its predicate chain is solid, and its post-market plan is sufficient. Here’s how to stress-test that belief.
For Founders and R&D Leaders
What failure mode is plausible even if testing hasn’t shown it? Think mechanistically about your device’s interaction with tissue, with time, with the real-world conditions of use. Your bench tests simulate idealized conditions. What happens under worst-case conditions — the heaviest patient, the smallest implant size, the longest implantation duration, the most aggressive surgical technique, the highest-humidity storage environment?
Are you testing for the harms your device could cause, or only for the performance it’s supposed to deliver? It’s natural to design verification and validation testing around your intended use. But the most dangerous failure modes are often orthogonal to intended use. Pelvic mesh worked for its intended purpose — structural support — while simultaneously eroding through tissue. The Therac-25 delivered correct doses most of the time while harboring a race condition that could deliver lethal doses under specific input sequences. Design your testing to hunt for what could go wrong, not just to confirm that things go right.
Who has the authority to stop or pause the program if a signal emerges? Name that person. Give them a title, a reporting line, and explicit executive backing. If the answer is “well, we’d all discuss it as a team,” you don’t have a safety governance system — you have a consensus process that will default to continuation under commercial pressure.
What would the FDA demand if they knew about your product’s plausible failure modes? Don’t wait for a 510(k) reviewer to ask the hard question. If your device uses a novel material in a new anatomical site, you can bet someone will eventually ask whether you’ve demonstrated safety in that specific context. If your device relies on software for a safety-critical function, someone will eventually ask about your software verification and validation, your hazard analysis, and your redundancy architecture. Preempt these questions. Generate the evidence voluntarily. It’s cheaper to run the study now than to manage the recall later.
Are you building a device submission or a safety system? The 510(k) or PMA submission is a regulatory event. It happens once (or periodically, for supplements). A safety system is continuous. It includes the people, processes, data streams, and decision triggers that will govern your product’s risk profile for as long as it’s on the market — potentially decades, for implantable devices. The submission gets you to market. The safety system keeps you there without destroying patients and shareholder value.
For Board Members and Investors
Is safety managed as a governance system or just a testing program? Does the company have routine safety review meetings with independent oversight? Are there pre-defined escalation criteria for adverse event trends? Is there a person or committee with explicit authority to halt distribution if signals warrant it? Or does safety only get discussed when a regulatory filing is due?
Active surveillance or passive hope? Ask what the company’s post-market surveillance plan includes beyond MAUDE reporting. Are they participating in or sponsoring registries? Do they have scheduled follow-up protocols for implant patients? Are they monitoring complaint trends with statistical rigor, or just filing individual reports? If the only post-market safety plan is “we’ll review adverse event reports,” that’s a red flag.
Does the predicate chain hold up to scrutiny? If the product was cleared via 510(k), trace the predicate chain. How many generations of predicates separate this product from a device that underwent independent clinical safety testing? Has the intended use, the anatomical site, or the patient population shifted meaningfully across the chain? A predicate chain is only as strong as its weakest link, and a chain that crosses anatomical sites or patient populations has very weak links indeed.
Is engineering (and safety expertise) embedded from the start? Did the company involve biocompatibility specialists, human factors engineers, and safety systems engineers in the concept and design phase? Or are they scrambling to add safety data after a reviewer or investor asked an uncomfortable question? Early engagement prevents investing in a fundamentally unsafe design. Late engagement means the surprise is already baked in — you just haven’t found it yet.
Remember: markets don’t punish risk; they punish negligence. Plenty of devices have known risks and succeed commercially — joint implants wear out, pacemaker leads fracture, surgical tools have learning curves. What destroys value is the revelation that the company knew about a risk and didn’t manage it — or worse, didn’t build the infrastructure to see it in the first place. When you do due diligence, a solid safety infrastructure isn’t a cost center; it’s a valuation protector.
The System Still Leaks: What’s Coming Next
The historical cases are instructive, but the future threat landscape for device safety is evolving in ways that will stress the existing infrastructure even further.
AI/ML-enabled devices introduce non-static risk profiles. A locked algorithm is one thing — but devices that learn or adapt over time can drift from their validated performance envelope without anyone noticing. The FDA’s predetermined change control plan (PCCP) framework is an attempt to address this, but it’s early-stage. Companies developing AI-enabled devices need safety monitoring systems that can detect algorithmic drift in real time — not just at periodic review intervals.
Cybersecurity is now a patient safety issue. Connected devices — insulin pumps, pacemakers, infusion systems — can be compromised in ways that directly threaten patient health. This is not hypothetical: the FDA has issued safety communications about cybersecurity vulnerabilities in specific insulin pumps and cardiac devices. The diagnostic infrastructure for cybersecurity-related device safety is nascent. Most device companies treat cybersecurity as an IT problem rather than a patient safety problem, which means the accountability structures aren’t in place.
Global supply chain complexity creates hidden material risks. The Philips CPAP case involved a foam material whose degradation behavior wasn’t adequately characterized. As device supply chains become more global and more complex, the number of materials, components, and subassemblies that could introduce latent risk increases. Companies that don’t have rigorous incoming material characterization, supplier qualification, and long-term material stability monitoring are building products on foundations they don’t fully understand.
Combination products blur regulatory boundaries. Devices that incorporate drugs (drug-eluting stents, antibiotic-impregnated implants), biologics (tissue-engineered products), or digital therapeutics create jurisdictional complexity. Which center at the FDA has primary oversight? What testing paradigm applies? The risk of falling through regulatory cracks — of no single authority owning the full risk profile — is elevated for combination products.
Bottom Line
The Dalkon Shield, the Therac-25, the DePuy ASR, pelvic mesh, the Philips CPAP — these were not unpredictable tragedies. They were system failures that allowed known or knowable dangers to persist because the diagnostic infrastructure to see the risk, or the accountability infrastructure to act on it, was absent or broken.
Safety isn’t a testing program. It isn’t a regulatory submission. It isn’t a checkbox. It’s a system — a continuously operating governance system that connects risk science to decision authority with enforceable accountability. And like any system, it works only when someone is truly responsible for its outcomes.
If your device safety strategy depends on “good engineers eventually catching problems” or “the FDA will tell us if something’s wrong,” it is brittle and will fail under pressure. The only durable approach is to build the structure that ensures plausible risks are investigated, safety signals are escalated, and someone with authority acts on them — before patients are harmed.
In the end, every successful medical device is a device that’s safe enough for people to benefit from — over years, over decades, in the messy reality of diverse patient bodies and unpredictable clinical environments. Ensuring that is a responsibility that cannot be delegated to predicates, check-the-box testing, or hope. It requires infrastructure, governance, and above all, accountability.
References
- Hines, J. Z., Lurie, P., Yu, E., & Wolfe, S. (2010). Left to their own devices: Breakdowns in United States medical device premarket review. PLOS Medicine, 7(7), e1000280.
- Zuckerman, D. M., Brown, P., & Nissen, S. E. (2011). Medical device recalls and the FDA approval process. Archives of Internal Medicine, 171(11), 1006–1011.
- U.S. Food and Drug Administration. (2019, April 16). FDA takes action to protect women’s health, orders manufacturers of surgical mesh intended for transvaginal repair of pelvic organ prolapse to stop selling all devices.
- Leveson, N. G., & Turner, C. S. (1993). An investigation of the Therac-25 accidents. Computer, 26(7), 18–41.
- Leveson, N. G. (2017). The Therac-25: 30 Years Later. Computer, 50(11), 8–11.
- Langton, D. J., Jameson, S. S., Joyce, T. J., et al. (2010). Early failure of metal-on-metal bearings in hip resurfacing and large-diameter total hip replacement. Journal of Bone and Joint Surgery (British Volume), 92-B(1), 38–46.
- Cohen, D. (2011). How safe are metal-on-metal hip implants? BMJ, 342, d3205.
- U.S. Food and Drug Administration. (2021, September). Philips Respironics recall of certain ventilators, BiPAP machines, and CPAP machines.