Why Medical Devices Fail FDA Review
How Investors Interpret Pathway Assumptions
February 27, 2026FDA Rejection Is Rarely About Bad Data
March 3, 2026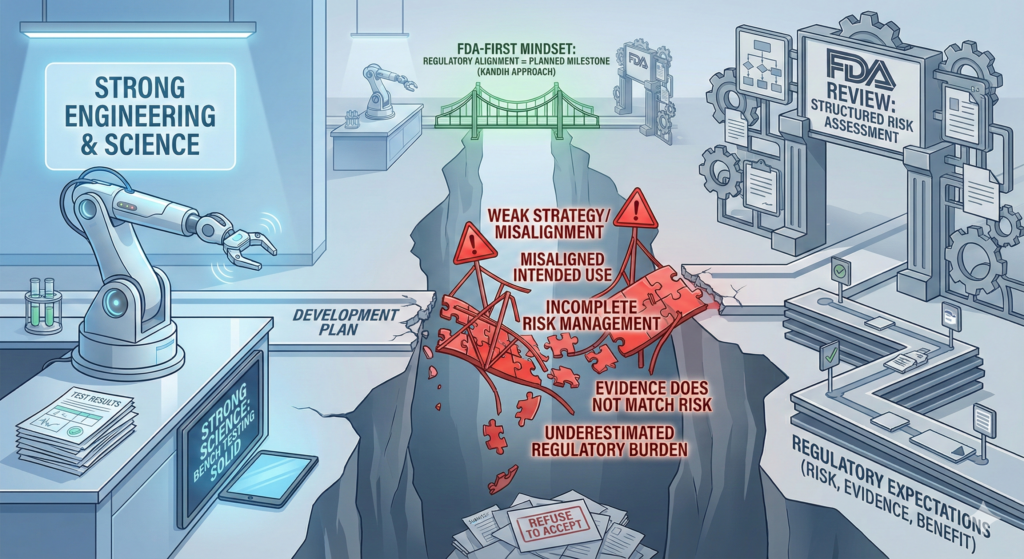
Direct answer: most medical devices do not fail FDA review because the science is weak. They fail because the strategy was weak.
In many cases, the engineering works. The bench testing looks solid. The prototype performs as expected.
But the submission fails because the development plan was not aligned with how the U.S. Food and Drug Administration evaluates risk, evidence, and benefit.
FDA review is not a science fair. It is a structured risk assessment process.
When companies misunderstand that, they stumble.
The Real Reasons Devices Fail FDA Review
1. Misaligned Intended Use
One of the most common failure points is unclear or overreaching intended use language.
If intended use:
Expands beyond predicate scope
Introduces new clinical claims
Targets a different patient population
FDA may determine the pathway is incorrect or the evidence insufficient.
This is not a scientific failure. It is a strategic one.
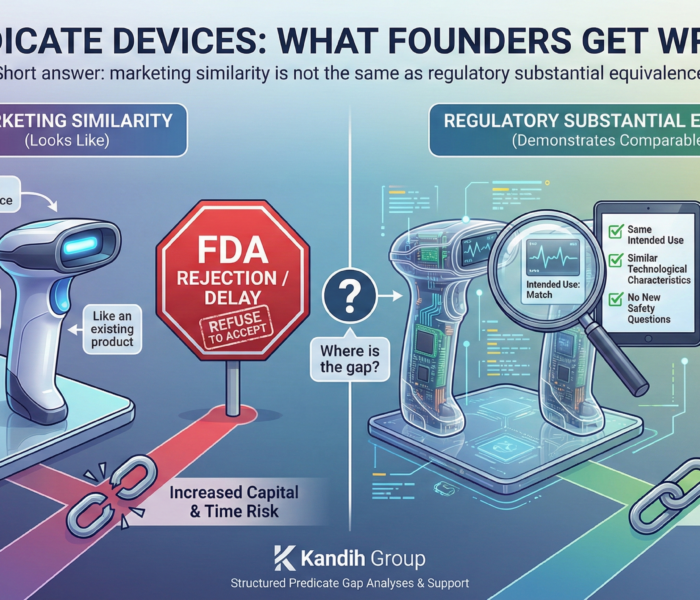
2. Weak Substantial Equivalence Arguments
For 510(k) submissions, failure often occurs because:
Technological differences were underestimated
New safety questions were not addressed
Risk mitigation was poorly documented
If substantial equivalence is not clearly demonstrated, clearance will not happen.
Again, this is not about whether the device works. It is about whether the risk comparison is defensible.
3. Incomplete Risk Management
FDA expects clear traceability:
Identified risks
Control measures
Verification testing
Validation data
If risk analysis is superficial or disconnected from testing, the submission looks fragmented.
FDA does not approve disconnected data.
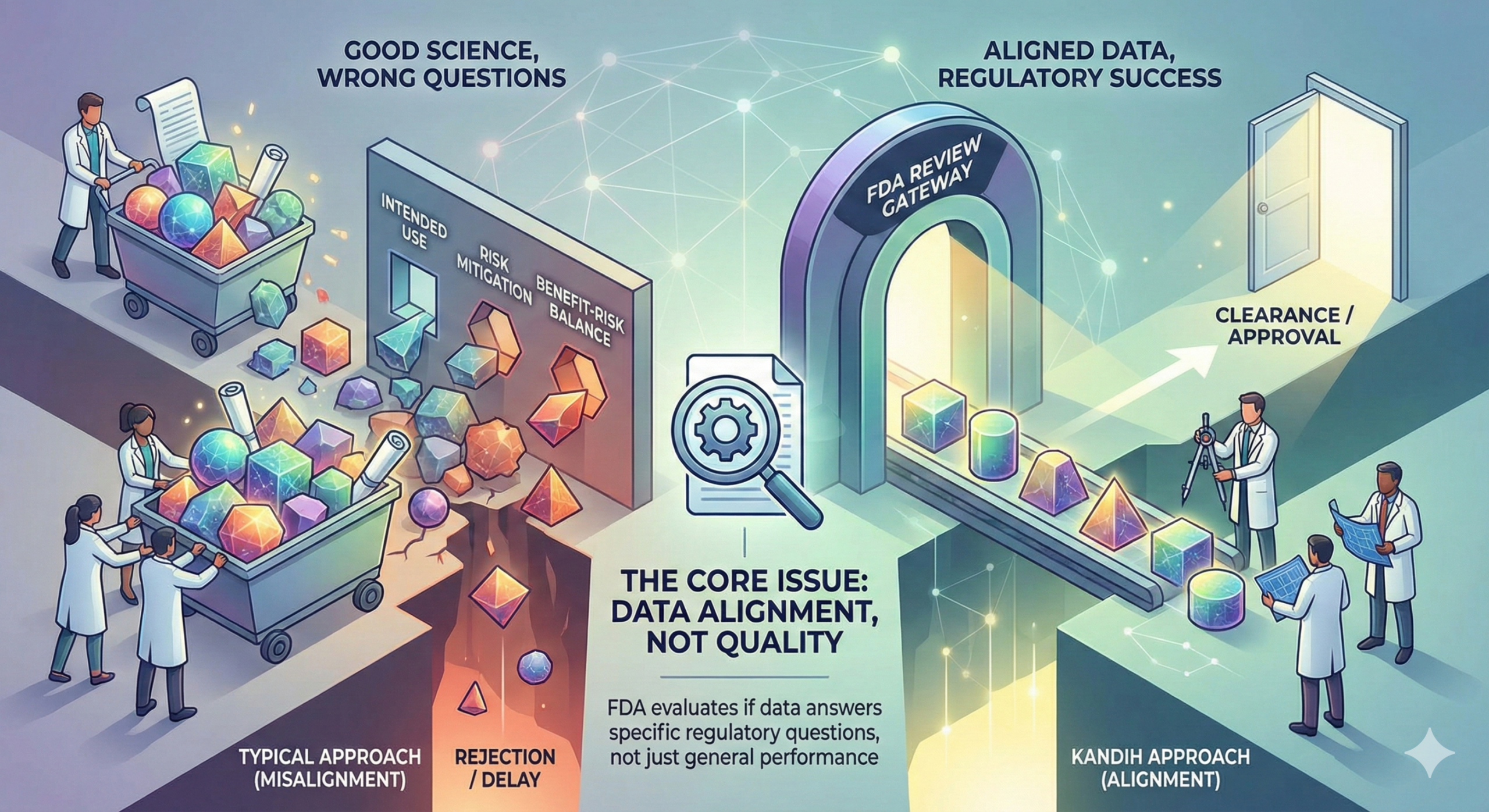
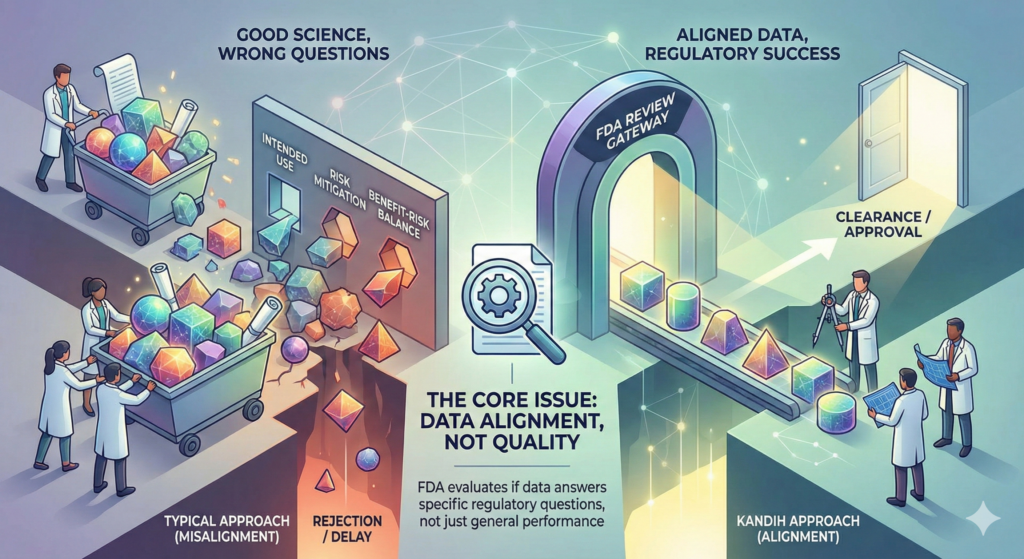
4. Evidence That Does Not Match the Risk Profile
Sometimes companies generate a large amount of data—but the wrong type of data.
Common issues include:
Bench testing that does not address identified hazards
Human factors studies that do not reflect real-world use
Clinical endpoints that do not support the claimed benefit
This creates review delays, additional information requests, or pathway escalation.
5. Overconfidence in “Innovation”
Innovation does not reduce regulatory burden. In many cases, it increases it.
Novel materials, AI-driven features, expanded claims, and new delivery mechanisms all introduce new safety questions.
If the regulatory strategy did not evolve alongside the innovation, review friction increases.
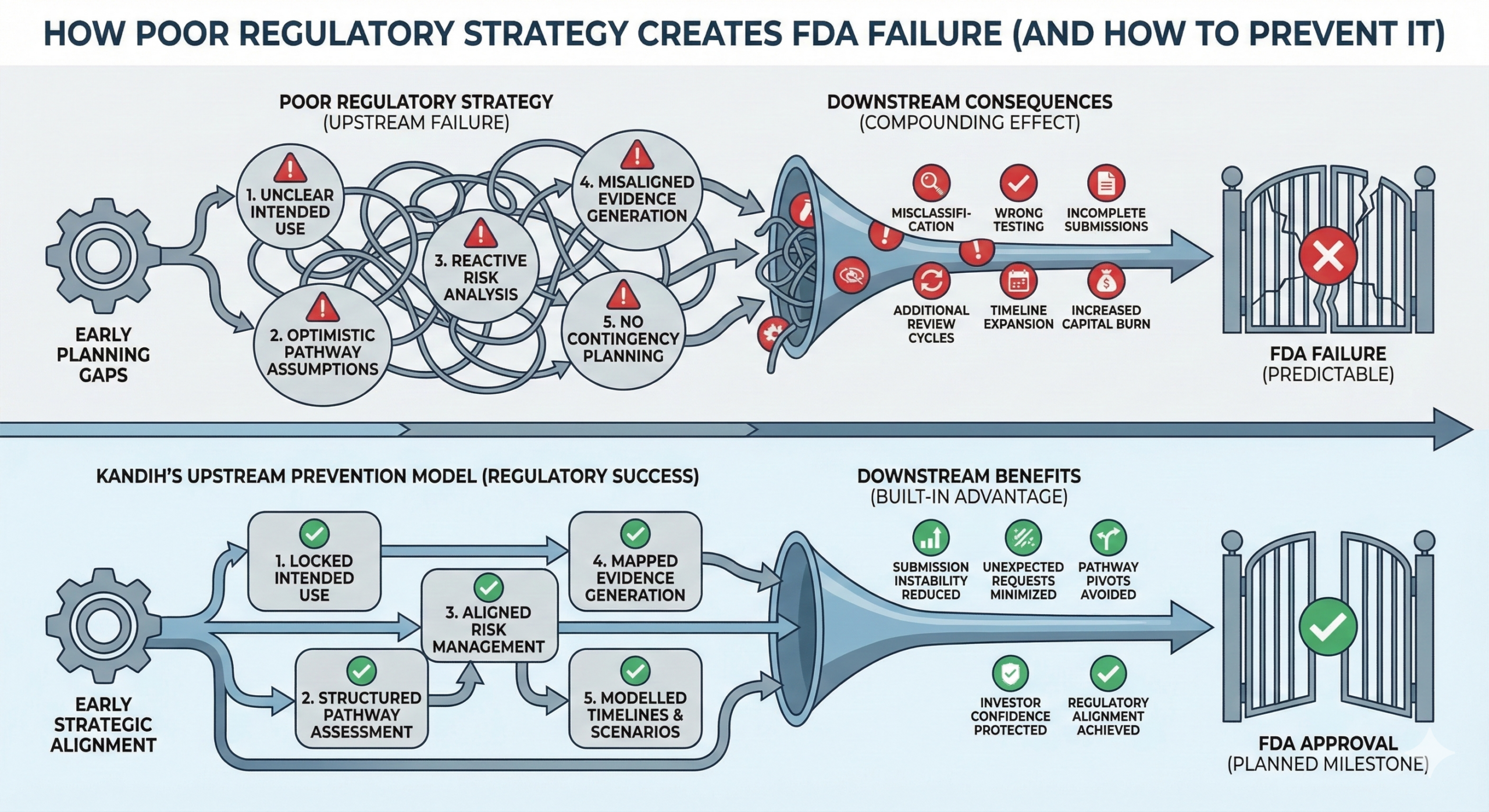
The Pattern Behind FDA Failure
When devices fail review, the pattern is usually:
Pathway chosen optimistically
Predicate assumptions stretched
Risk underestimated
Evidence generated reactively
Submission assembled defensively
That is a strategic breakdown.
Scientific problems can often be solved.
Strategic misalignment compounds.
What FDA Actually Evaluates
FDA reviewers ask structured questions:
Is the device properly classified?
Does the intended use align with the pathway?
Are risks identified and mitigated?
Does the evidence match the risk profile?
Is the benefit-risk balance supported?
If the answers are unclear, review slows.
If the logic is inconsistent, review stops.
Where Kandih Comes In
This is where Kandih Group applies an FDA-first development philosophy.
Kandih supports companies by:
Building regulatory strategy before design decisions harden
Aligning intended use with realistic classification
Conducting structured risk and pathway analyses
Mapping evidence generation directly to risk mitigation
Stress-testing submissions before FDA does
Instead of building a device and then defending it, teams develop in alignment with FDA logic from day one.
That reduces:
Refuse-to-Accept decisions
Multiple review cycles
Forced pathway pivots
Investor confidence erosion
Bottom Line
Medical devices rarely fail FDA review because they lack science.
They fail because the development strategy did not match regulatory expectations.
Strong engineering is not enough.
Regulatory alignment determines survivability.
An FDA-first mindset turns regulatory review from a hurdle into a planned milestone.
References
FDA – Refuse to Accept Policy for 510(k)s
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/refuse-accept-policy-510ks
FDA – Design Controls for Medical Devices
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/design-controls-medical-devices
FDA – Substantial Equivalence in Premarket Notifications (510(k))
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/substantial-equivalence-premarket-notifications-510k
FDA – Premarket Approval (PMA)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma

{kind=link}
{kind=link}
{kind=link}