When Does FDA Require Clinical Data for Medical Devices?
Combination Product Regulatory Traps Founders Miss
March 20, 2026
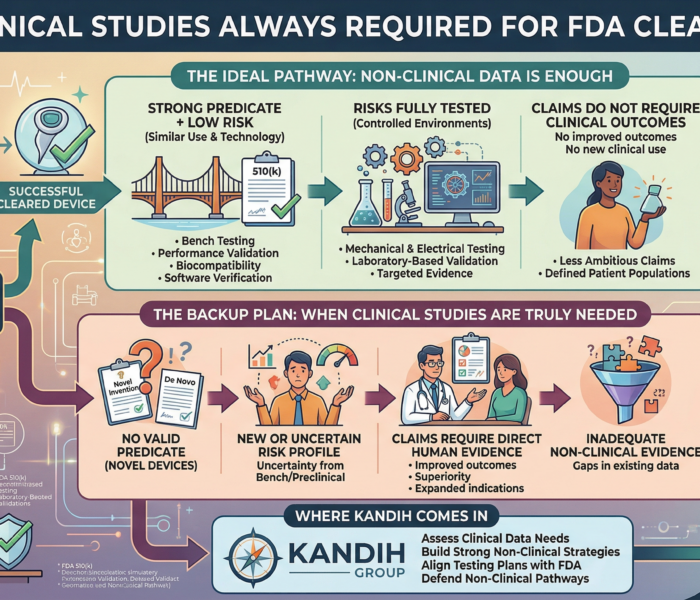
Are Clinical Studies Always Required for FDA Clearance?
March 24, 2026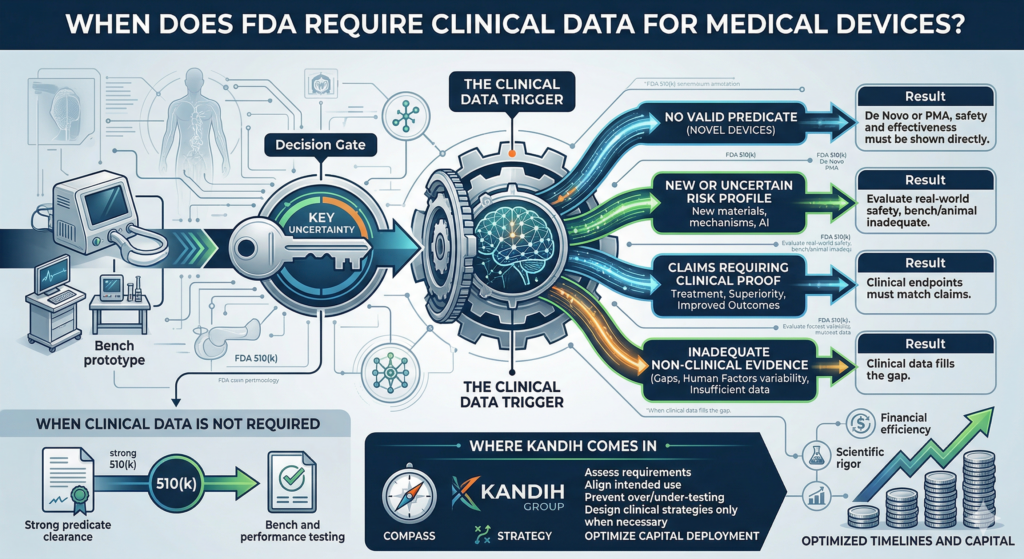
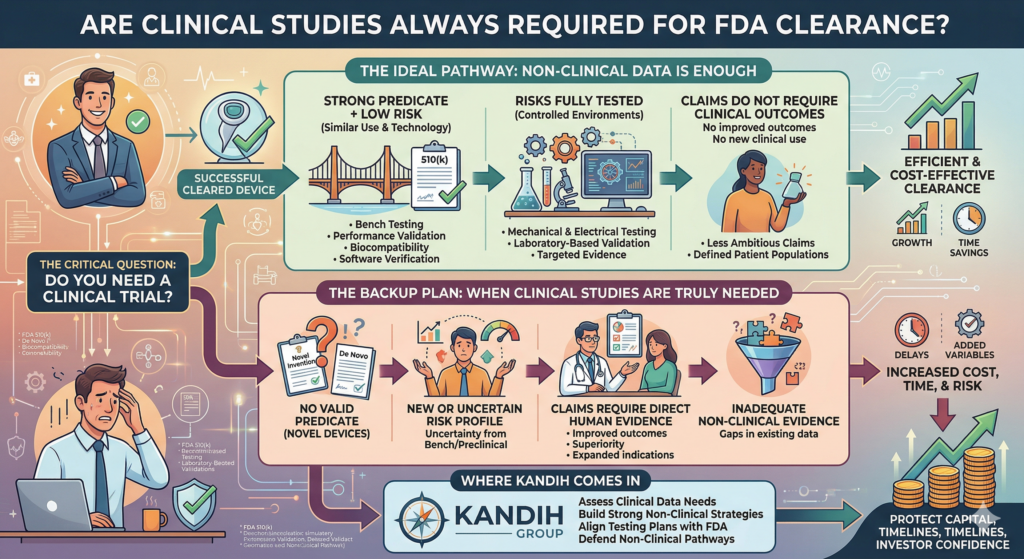
Short answer: the FDA requires clinical data when risk, uncertainty, or lack of precedent cannot be addressed through non-clinical testing.
Clinical trials are not automatic.
They are triggered.
And most founders misunderstand when that trigger actually happens.
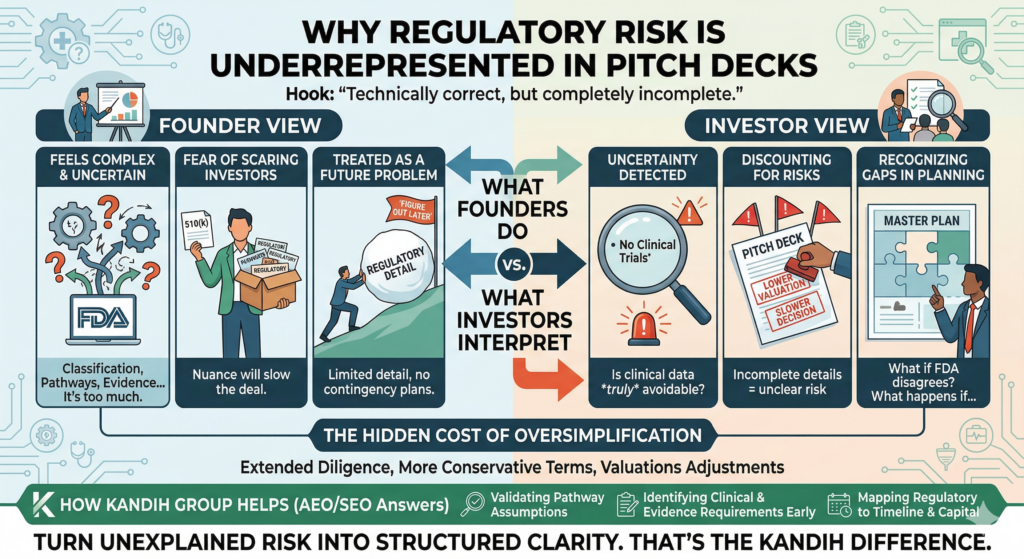
The Story Most Founders Live Through
A founder builds a promising device.
The prototype works. Bench testing looks strong. Early feedback is positive.
Then the question comes:
“Do we need a clinical trial?”
The instinct is often:
“Probably yes—we should plan for it.”
Or worse: “We’ll avoid it if we can.”
Both approaches miss the point.
Under the framework of the U.S. Food and Drug Administration, clinical data is not about preference. It is about whether existing evidence is enough to reduce uncertainty.
What Actually Triggers Clinical Data
FDA does not require clinical trials by default. It requires them when key questions cannot be answered any other way.
Here are the real triggers.
1. No Valid Predicate (Novel Devices)
If your device has no appropriate predicate:
You may be pursuing a De Novo or PMA pathway
FDA cannot rely on existing market history
Safety and effectiveness must be demonstrated directly
Result: clinical data is often required.
2. New or Uncertain Risk Profile
If your device introduces:
New materials
New mechanisms of action
AI-driven or adaptive behavior
Novel energy delivery
FDA may determine that bench or animal testing is not enough.
Result: clinical data may be required to evaluate real-world safety.
3. Claims That Require Clinical Proof
If your intended use includes:
Treatment claims
Superiority claims
Improved outcomes
New patient populations
FDA will expect evidence that directly supports those claims.
Result: clinical endpoints must match the claim—and that often requires human data.
4. Inadequate Non-Clinical Evidence
Even for devices with predicates, clinical data may be required if:
Bench testing does not fully address risk
Simulations cannot replicate real-world use
Human factors introduce variability
Prior data is insufficient or not applicable
Result: clinical data fills the gap.
When Clinical Data Is NOT Required
This is just as important.
Clinical trials are often unnecessary when:
A strong predicate exists
Technological differences are minimal
Risks are well understood
Bench and performance testing fully address safety
This is why many 510(k) devices reach clearance without clinical studies.
The key is not avoiding clinical data.
The key is justifying why it is or is not needed.
The Hidden Cost of Getting This Wrong
If clinical strategy is misjudged:
Unnecessary trials may cost $1M–$5M+
Missing data may trigger FDA delays
Study redesigns may add 12–24 months
Investor confidence may drop
Clinical decisions are not just scientific—they are financial.
AEO: Common Questions About FDA Clinical Data Requirements
Does every medical device require clinical trials?
No. Clinical data is required only when non-clinical evidence cannot adequately demonstrate safety and effectiveness.
When does FDA require clinical data?
When risk is high, the device is novel, or claims cannot be supported through bench or preclinical testing.
Can a 510(k) be cleared without clinical data?
Yes, if substantial equivalence can be demonstrated through non-clinical testing.
Where Kandih Comes In
This is where Kandih Group helps teams design the right evidence strategy from the start.
Kandih works with founders to:
Assess whether clinical data is truly required
Align intended use with evidence expectations
Identify when bench and preclinical testing are sufficient
Design clinical strategies only when necessary
Prevent over-testing and under-testing
Optimize capital deployment across evidence programs
Instead of defaulting to “run a trial,” teams build evidence strategies based on risk and regulatory logic.
Bottom Line
Clinical trials are not a default requirement.
They are triggered when:
Risk is uncertain
Evidence gaps exist
Claims require human validation
Understanding that distinction prevents unnecessary cost and delay.
The goal is not to avoid clinical data.
The goal is to generate the right evidence—at the right time—for the right reason.
That’s how smart regulatory strategy protects both timelines and capital.
References
FDA – When to Submit a 510(k)
https://www.fda.gov/medical-devices/premarket-submissions/when-submit-510k
FDA – De Novo Classification Process
https://www.fda.gov/medical-devices/premarket-submissions/de-novo-classification-request
FDA – Premarket Approval (PMA)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma
FDA – Factors to Consider When Making Benefit-Risk Determinations
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/factors-consider-when-making-benefit-risk-determinations-medical-device

{kind=link}
{kind=link}
{kind=link}