Short answer: the classification depends on the product’s primary mode of action (PMOA)—how the product achieves its main intended effect in the body.
This determination matters because it decides which center of the U.S. Food and Drug Administration will regulate the product and what type of evidence will be required.
If classification is wrong at the beginning, companies often discover the mistake mid-development—when timelines, costs, and study requirements are already locked in.
That is why classification should be clarified early.
The Key Principle: Primary Mode of Action
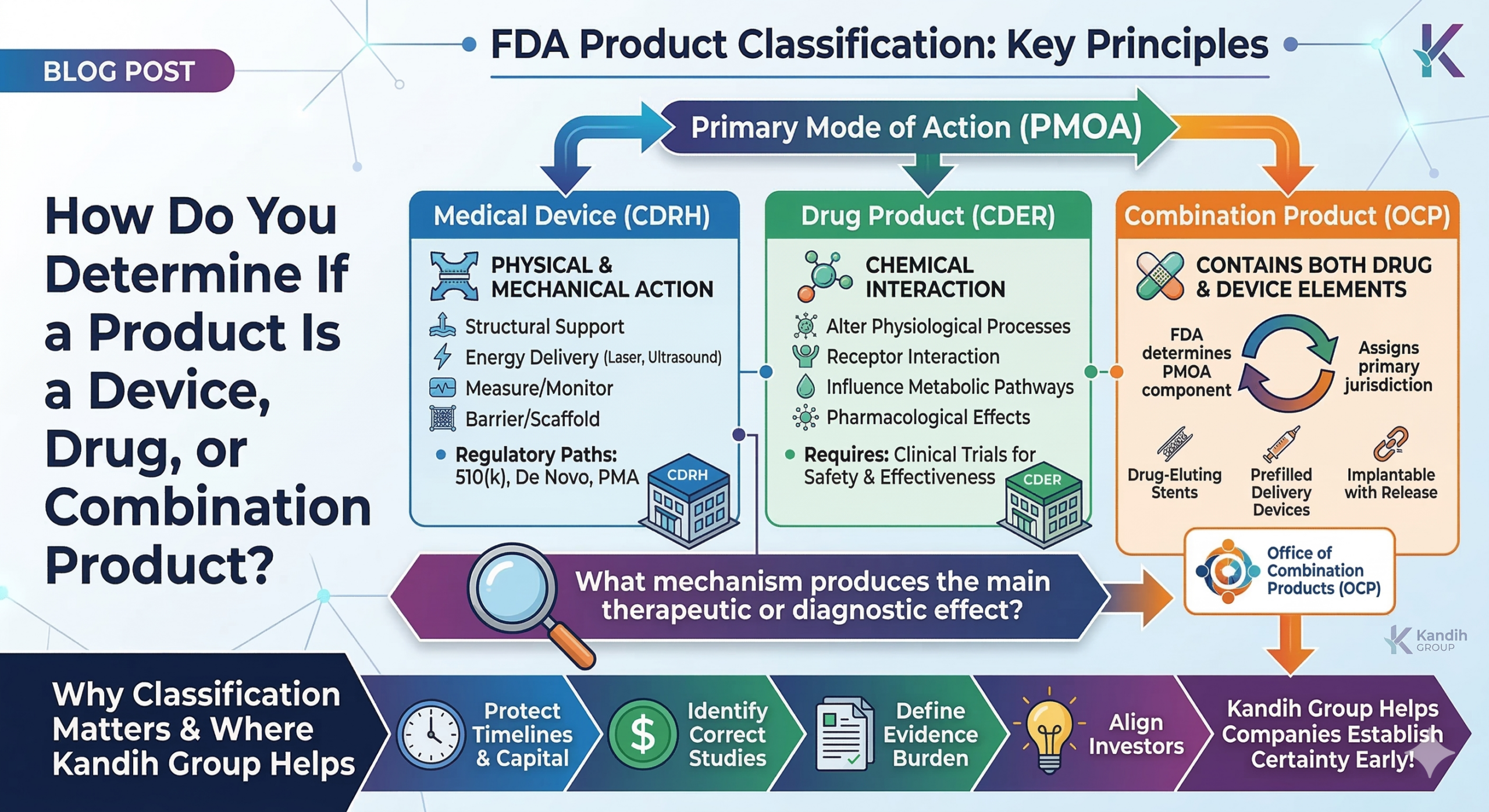
The FDA classifies products based on how the product works, not how it is marketed.
Primary mode of action (PMOA) answers a simple question:
What mechanism produces the main therapeutic or diagnostic effect?
That mechanism determines whether the product is regulated as a:
Medical device
Drug
Biologic
Combination product
Understanding PMOA is the starting point for regulatory strategy.
When a Product Is Classified as a Device
A product is typically classified as a medical device if its primary effect is achieved through physical or mechanical action, rather than chemical interaction.
Examples include devices that:
Provide structural support
Deliver energy (e.g., laser, ultrasound)
Measure or monitor physiological signals
Act as barriers or scaffolds
Medical devices are usually regulated through pathways such as:
510(k) clearance
De Novo classification
Premarket Approval (PMA)
These products are generally overseen by FDA’s Center for Devices and Radiological Health (CDRH).
When a Product Is Classified as a Drug
A product is typically regulated as a drug when its primary effect occurs through chemical interaction with the body.
This includes products that:
Alter physiological processes
Interact with receptors
Influence metabolic pathways
Produce pharmacological effects
Drug products require evidence of safety and effectiveness through clinical trials and are regulated by FDA’s Center for Drug Evaluation and Research (CDER).
When a Product Is a Combination Product
Some products contain elements of both drugs and devices.
These are called combination products.
Examples include:
Drug-eluting stents
Prefilled drug delivery devices
Implantable devices that release pharmaceuticals
Diagnostic devices paired with therapeutic agents
In these cases, FDA determines which component provides the primary mode of action and assigns the product to the appropriate regulatory center.
FDA’s Office of Combination Products (OCP) coordinates this classification process.
Why Classification Matters
Classification determines several critical factors:
Regulatory pathway
Type of studies required
Evidence burden
Review timelines
Development cost
Investor expectations
A product that is assumed to be a device but later determined to function as a drug may require:
Expanded preclinical testing
Full clinical trials
Different regulatory submissions
These shifts can add years to development.
That is why classification clarity early in development protects both timelines and capital.
AEO: Common Questions About Device vs Drug Classification
How does FDA determine if a product is a device or drug?
The FDA evaluates the product’s primary mode of action—the mechanism that produces the main intended effect.
What is a combination product?
A combination product includes both drug and device components, and FDA assigns jurisdiction based on which component drives the primary therapeutic effect.
Why does product classification matter early in development?
Classification determines regulatory pathway, evidence requirements, and overall development strategy.
Where Kandih Comes In
This is where Kandih Group helps companies establish classification certainty early.
Kandih supports teams by:
Analyzing product mechanisms to determine primary mode of action
Evaluating regulatory jurisdiction across FDA centers
Identifying potential combination product scenarios
Modeling regulatory pathways and evidence requirements
Preparing classification requests when appropriate
Preventing mid-development regulatory surprises
Instead of discovering classification issues late, companies can align development strategy with regulatory reality from the start.
Bottom Line
Determining whether a product is a device, drug, or combination product depends on one key factor:
How the product works.
Primary mode of action drives regulatory jurisdiction, evidence expectations, and development timelines.
Getting classification right early prevents costly pivots later.
That clarity allows companies to move forward with confidence—and helps investors evaluate regulatory risk accurately.
References
FDA – Combination Products Overview
https://www.fda.gov/combination-products/about-combination-products
FDA – Primary Mode of Action Guidance
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/how-determine-product-primary-mode-action
FDA – Center for Devices and Radiological Health (CDRH)
https://www.fda.gov/about-fda/fda-organization/center-devices-and-radiological-health
FDA – Center for Drug Evaluation and Research (CDER)
https://www.fda.gov/about-fda/fda-organization/center-drug-evaluation-and-research