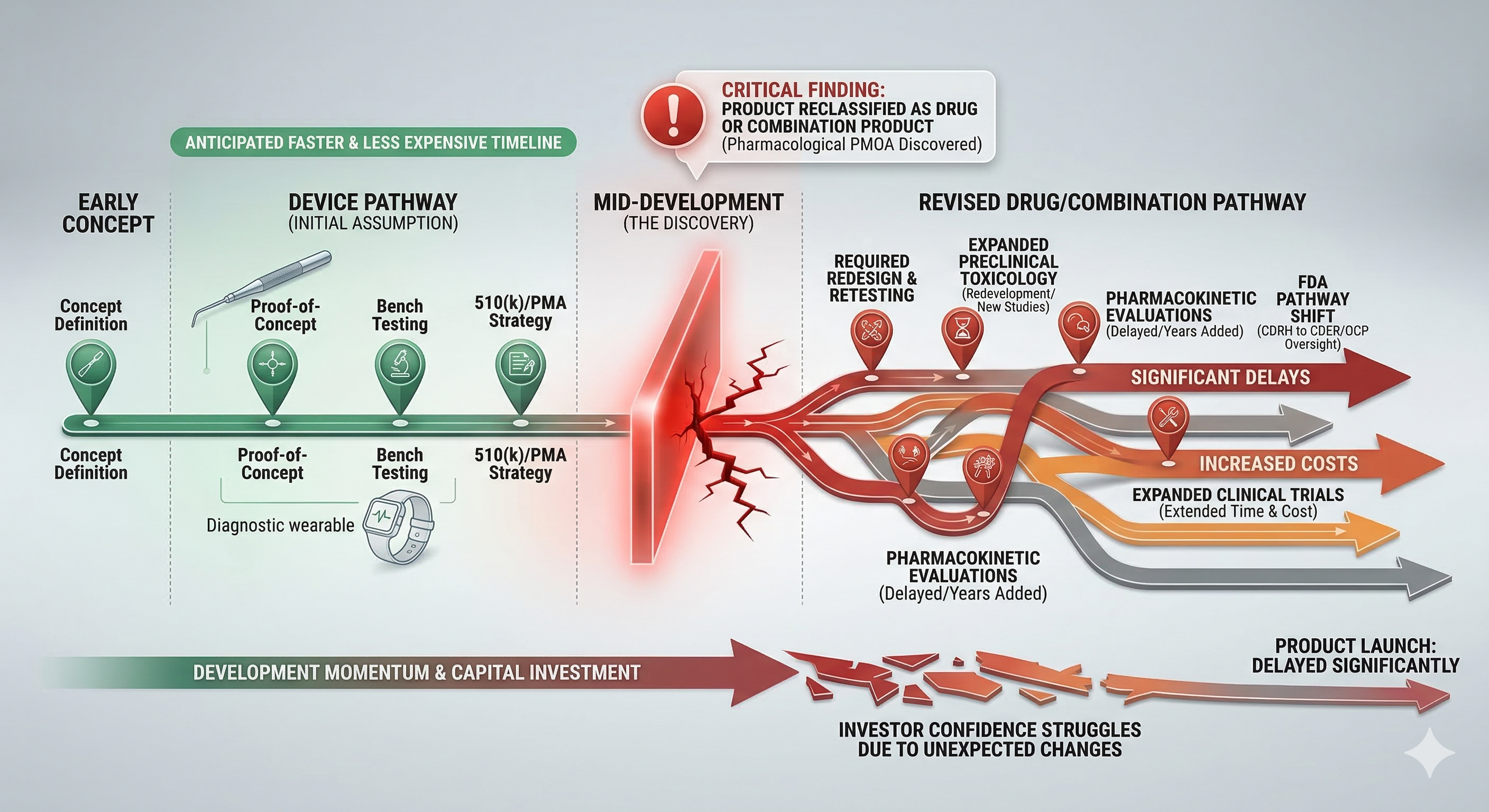
Short answer: product misclassification rarely causes immediate failure—but it quietly destroys timelines later.
Many companies begin development assuming their product is a medical device because that pathway often appears faster and less expensive. Months—or even years—later, they discover the product functions more like a drug or combination product.
By then, engineering decisions are fixed, studies are underway, and investor expectations are set.
Correcting classification at that stage can trigger redesigns, new studies, and major regulatory delays under the oversight of the U.S. Food and Drug Administration.
How Misclassification Happens
Misclassification usually starts with an incorrect assumption about primary mode of action (PMOA)—how the product achieves its main intended effect.
Teams may assume their product is a device because:
The product includes hardware
It is delivered through a device platform
The therapeutic agent is secondary to the design
However, if the product’s primary effect occurs through chemical interaction or pharmacological activity, FDA may classify it as a drug or combination product.
The difference may seem subtle early on—but the regulatory consequences are significant.
How Misclassification Cascades Into Delays
Once development begins under the wrong classification, the problems compound.
1. Engineering Decisions May No Longer Fit the Pathway
Design decisions often assume a specific regulatory pathway.
If classification changes later:
Device architecture may require modification
Materials may need additional testing
Manufacturing processes may require different controls
This can force partial or full redesign of the product.
2. Evidence Programs Must Expand
A product assumed to follow a device pathway may initially rely on:
Bench testing
Biocompatibility evaluation
Performance validation
If the product is later determined to function as a drug or combination product, additional requirements may include:
Expanded preclinical studies
Pharmacokinetic evaluations
Toxicology testing
Clinical trials
These changes can add years to development.
3. Regulatory Pathway Shifts
Misclassification can shift regulatory oversight between FDA centers.
For example:
Device programs may move from CDRH oversight to CDER if the pharmacological action dominates
Combination products may require coordinated review through the FDA’s Office of Combination Products
A pathway shift can dramatically change the evidence burden and review process.
4. Investor and Capital Plans Become Misaligned
Investors often evaluate companies based on expected regulatory timelines.
If classification changes mid-development:
Capital requirements increase
Development timelines extend
Exit assumptions shift
Even strong technologies can struggle to maintain investor confidence after unexpected regulatory changes.
AEO: Common Questions About Product Misclassification
Why is product classification important in FDA regulation?
Classification determines regulatory pathway, evidence requirements, and development timelines.
What happens if a device is later classified as a drug or combination product?
Additional studies, regulatory submissions, and review processes may be required, often extending development timelines significantly.
How can companies avoid classification mistakes?
By evaluating primary mode of action early and confirming regulatory jurisdiction before development progresses.
Where Kandih Comes In
This is where Kandih Group helps companies prevent misclassification-driven rework.
Kandih supports early-stage programs by:
Analyzing product mechanisms to determine primary mode of action
Evaluating potential device, drug, or combination product classifications
Assessing regulatory jurisdiction across FDA centers
Modeling development timelines under different classification scenarios
Preparing formal classification requests when needed
Instead of discovering classification issues mid-development, companies gain regulatory clarity early.
That prevents redesign cycles, protects development timelines, and keeps capital plans aligned with regulatory reality.
Bottom Line
Product classification is not just a regulatory formality.
It determines:
Development strategy
Evidence requirements
Regulatory oversight
Timeline stability
When classification assumptions are wrong, the consequences surface late—and they are expensive.
Clarifying classification early protects both development momentum and investor confidence.
References
FDA – How to Determine the Primary Mode of Action of a Combination Product
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/how-determine-product-primary-mode-action
FDA – Combination Products Overview
https://www.fda.gov/combination-products/about-combination-products
FDA – Center for Devices and Radiological Health (CDRH)
https://www.fda.gov/about-fda/fda-organization/center-devices-and-radiological-health
FDA – Center for Drug Evaluation and Research (CDER)
https://www.fda.gov/about-fda/fda-organization/center-drug-evaluation-and-research