Safety is Not Advice: Why Better Data Won’t Save Your Drug Without Diagnostic Infrastructure
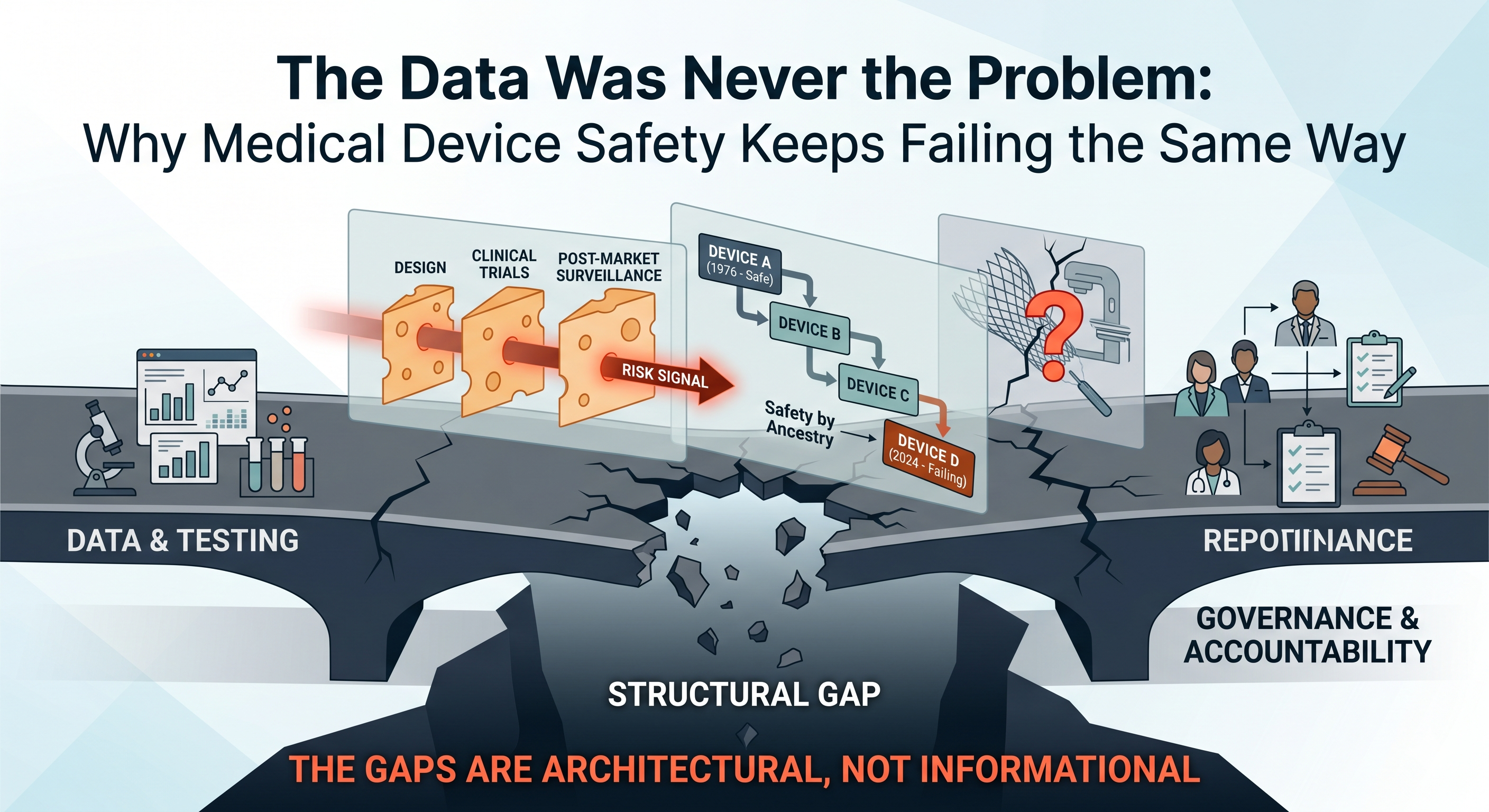
The Data Was Never the Problem: Why Medical Device Safety Keeps Failing the Same Way
April 22, 2026
Why 510(k) Submissions Get Delayed: 5 Toxicology Mistakes Founders Miss
April 24, 2026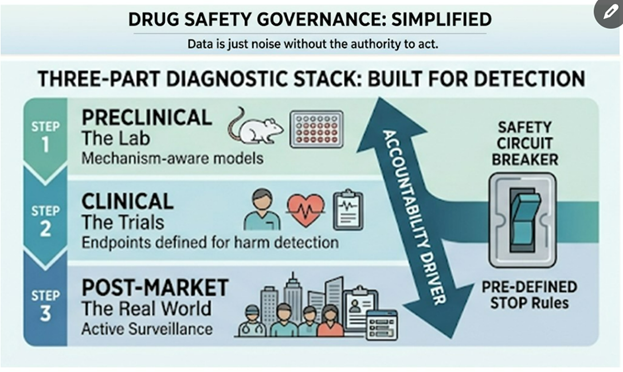
I design FDA evidence that protects capital · Regulatory Toxicologist, PhD DABT · Medical Devices · Diagnostics · Drugs · Biologics · Medical Foods · Founder & CEO, Kandih BioScience
Why drug safety failures keep happening—and why “better data” won’t save you without diagnostic infrastructure
The Predictable Anatomy of Drug Safety Disasters
Every major medical product safety disaster has a predictable anatomy. From thalidomide in the 1960s to Vioxx in the 2000s, from Avandia to recent issues with JAK inhibitors, ranitidine, and opioids, the pattern is the same. It’s not that the science was missing – often the safety signals were there – but the system refused to be accountable to the science. In each case, warning signs existed, yet no one owned the obligation to act on them. This uncomfortable truth underpins why simply getting “better data” isn’t enough: safety advice only matters when someone is accountable to enforce it.
“If It’s Risky, Toxicology Will Catch It” – A Costly Myth
Many founders assume that if a drug or therapy is risky, routine toxicology will automatically catch it. In reality, toxicology doesn’t “discover” risks in a vacuum – it depends on a diagnostic infrastructure spanning three layers of a drug’s journey:
- Preclinical Predictivity: Models and assays capable of detecting plausible harms in the lab (e.g. cell studies, animal models).
- Clinical Visibility: Trial designs, endpoints, and monitoring that are set up to observe those harms in human studies.
- Post-Market Detection: Surveillance systems to find real-world safety signals fast enough to matter.
If any one of these layers is weak, safety becomes a post-mortem exercise – figuring out causes only after patients are hurt. In other words, regulatory toxicology can catch risks only if the system is built to see them. History provides stark examples of how gaps in diagnostic infrastructure lead to tragedy.
Thalidomide: A Diagnostics Failure, Not a Science Failure
Thalidomide is often remembered as a “science” failure of the 1950s–60s, but it was really a failure of diagnostics and accountability. At the time, regulators did not require embryo-fetal developmental tests in new drug development; companies weren’t compelled to run them. Even if such tests had been done, thalidomide’s notorious teratogenic (birth defect–causing) effect is highly species-dependent – standard rodent studies didn’t reveal the limb deformities that appeared in rabbits and primates. In other words, “no signal” simply meant “no one built a test capable of seeing the risk.”
Under the false assumption of safety, thalidomide was widely given to pregnant women for nausea. The result: thousands of infants born with severe malformations before the alarm was finally raised. It took the world witnessing this devastation for regulators to change course – after the fact, requirements were established for more predictive developmental testing (including tests in multiple species). The lesson was painful: lack of a diagnostic infrastructure rendered the risk invisible until it was too late. Thalidomide wasn’t unpredictable; it was just undiagnosed by a system not accountable to find the truth early.
Vioxx: The Signal Was There – The System Looked Away
If thalidomide was a warning about missing tests, Vioxx (rofecoxib) was a warning about ignoring tests and mechanistic signals. Vioxx, a COX-2–selective anti-inflammatory drug, was launched in 1999 as a safer alternative to older pain relievers. But from the beginning, there was a plausible mechanism for cardiovascular harm: inhibiting COX-2 reduces prostacyclin (a blood vessel protectant) without affecting COX-1’s pro-clotting thromboxane – potentially tipping the body toward clot formation. This was known as the FitzGerald hypothesis, and even Merck’s own scientists raised concerns early on about a heightened risk of heart attacks and strokes.
What happened next is telling. The clinical program wasn’t designed to properly detect cardiovascular risk. The trials used for FDA approval were short, enrolled low-cardiac-risk patients, and focused on Vioxx’s GI benefits (reducing ulcers) rather than heart safety. None of the studies had a standardized procedure to collect or adjudicate cardiac events. In essence, the trial architecture made the potential harm hard to see. And once the drug was on the market, post-market surveillance was the usual passive reporting system – slow, noisy, and too late. After approval, busy clinicians seeing heart attacks in an older patient would not immediately tag Vioxx as the culprit; a heart attack doesn’t come with a label saying “caused by arthritis medication.” Passive pharmacovigilance relies on someone connecting dots that the system itself should connect automatically. As a result, early warning signs were missed or dismissed for years while Vioxx was aggressively marketed as safe.
This wasn’t a gap in data; it was a gap in accountability for signal governance. The safety signal existed (higher rates of cardiovascular events in Vioxx users) – but no one in the system had both the clear sight and the authority to pump the brakes. Merck’s internal teams saw hints of risk, the FDA had questions, some clinicians had suspicions – yet Vioxx stayed on the market until 2004, when definitive trial data finally forced a withdrawal. In retrospect, multiple independent analyses concluded that Vioxx’s dangers could have been recognized much sooner if either the trials or the surveillance had been structured to catch the signal[5]. The passive FDA Adverse Event Reporting System (AERS) simply wasn’t robust enough, hampered by underreporting and difficulty attributing common events like heart attacks to a drug[6]. The core issue was that safety insights were treated as advice, not as orders – and advice can be (and was) ignored in favor of revenue and optimism.
The Core Problem: Science vs. Decision Rights
These cases reveal a fundamental truth: the science often flags risks, but the people who see the risk don’t always have the power to act on it. In large programs, a toxicologist might raise concerns, a clinician might report an odd adverse event, or a regulator might ask pointed questions – but unless someone in the chain has the decision rights explicitly tied to safety outcomes, those warnings can die on the vine. Organizations without clear safety governance will naturally drift toward speed, marketability, and profit. In such an environment, safety advice becomes optional.
In the Vioxx saga, for example, Merck scientists’ concerns about clotting risk did not translate into a halted program or a redesigned trial – because the momentum of a blockbuster drug in development carried the team past those concerns. No single role had the mandate to say “this mechanism is dangerous; stop or radically change the plan.” When safety decision-making is diffuse, the default trajectory is to push forward and hope for the best. This is why we keep seeing the same failure mode: the people who recognize the risk aren’t empowered to rein in the program.
Bottom line – the problem isn’t a lack of scientific insight; it’s the lack of accountability to that insight. Good science without enforcement is like a fire alarm in an empty building. To prevent disasters, someone must not only ring the alarm, but also have the authority to evacuate the building.
Diagnostic Infrastructure: The Missing Asset Class
How do we make safety more than just advice? By treating diagnostic infrastructure as an operational asset – as core to the program’s success as the product’s own IP. If you’re a biotech founder, you might think of diagnostics as just “tests” you run to check a box. If you’re an investor, you might think of it as simply “data” to be collected for regulatory filings. If you’re a regulator, you consider it “evidence” to base decisions on. All true, but incomplete. Diagnostic infrastructure is the machinery that turns risk science into enforceable behavior.
Think of it as a system-within-the-system that defines how and when we detect danger, and what we do about it. It includes choices like:
- Which nonclinical models you use (and why those models are relevant to your drug’s mechanisms).
- How clinical endpoints are defined and adjudicated – are you only measuring outcomes that make your drug look good, or also those that might reveal harm?
- Whether trials include high-risk populations who might show problems sooner (versus only healthy or low-risk subjects).
- Whether monitoring is designed for mechanistic harms – e.g. if your drug might affect the heart, did you include cardiac imaging or biomarkers, or just hope nothing happens?
- How post-market signals are captured: do you rely purely on passive reporting, or do you have active surveillance (such as scheduled follow-up studies, patient registries, or data-mining of electronic health records)?
- How quickly safety actions are triggered: is there a predefined threshold of risk at which you pause dosing or adjust a trial, or do you debate each time whether it’s “real”?
- Who ultimately has authority to pause, escalate, or redesign the program when a signal emerges.
Without this infrastructure, even the best toxicology findings or safety signals become nothing more than memos – easy to ignore. A fancy preclinical assay might show a worrying result, but if no process forces a team to address it (redesign the molecule, add a safety study, etc.), it remains academic knowledge. Many startups and even big pharma teams run what they call “safety programs,” but in reality these are often just hope-based checklists: run the standard battery of tests, tick the boxes, and proceed unless a catastrophe leaps out. True safety management means building a system that actively seeks out potential harm and compels the organization to deal with it.
The Regulatory Toxicologist’s Real Job: Enforcing Accountability
People often view regulatory toxicology as a compliance function – the team that runs required studies and files reports to satisfy the FDA or EMA. But in a truly safety-centric operation, the regulatory toxicologist (and the safety team broadly) should be the governance engine for safety uncertainty. Their role is not just to generate data, but to ensure that data leads to decisions. A strong regulatory toxicologist on a development team will:
- Map plausible harms early. They proactively ask: based on this drug’s mechanism, what organs or systems could be affected? What would that look like (tissue changes, clinical symptoms), and how could we monitor for it? (Mechanism → target organ/tissue → clinical phenotype → monitoring plan).
- Design detection into studies. Instead of simply running the minimum required studies, they influence study design to include measurements that would catch the predicted risks. (For instance, if liver toxicity is a concern, ensure liver enzymes are checked frequently and criteria are in place to stop if they rise.)
- Build clear escalation criteria. They define in advance what happens if a safety signal appears. What level of enzyme elevation, what frequency of adverse event, etc., triggers a dose reduction, a trial hold, additional investigations, or termination? By pre-defining these “stopping rules” or redesign triggers, they remove ambiguity and prevent the team from moving the goalposts when a problem arises.
- Align evidence with regulatory action. They think like regulators: what evidence will the FDA or EMA consider a real signal versus a fluke? They then push to gather that kind of evidence early. This might mean conducting a specific long-term study or including a certain high-risk patient group, so that down the road the company isn’t caught saying “we have signals, but no definitive data.” In other words, generate the evidence that will force a regulatory response (if the risk is real) rather than evidence that can be waved away.
- Protect the program’s value by preventing late-stage blowups. A good toxicologist knows that nothing tanks a biotech’s valuation like a safety fiasco in Phase III or post-approval withdrawal. By catching and resolving issues in Phase I or II, they are not just saving lives but also preserving the company’s investment. This often means making unpopular recommendations to slow down or run an extra study – but an early pause is far better than a catastrophic failure after pouring in millions of dollars.
If your team isn’t doing these five things, you don’t truly have a safety program – you have a “hope and proceed” strategy. Relying on good intentions and basic check-the-box tests will eventually fail. As one might say, hope is not a strategy. Regulatory toxicology done right is about inserting accountability at every step of R&D – it’s the safety circuit-breaker for the project.
The System Still Leaks: Modern Examples
Lest anyone think these issues are purely historical, recent events show that our diagnostic stack still has holes – and still needs stronger accountability. Consider these modern parallels:
- Avandia (Rosiglitazone) and Diabetes Drugs: In the 2000s, the diabetes drug Avandia was found to increase heart attack risk, triggering a firestorm of controversy. Only after this crisis did regulators insist that every new type 2 diabetes medication undergo a dedicated cardiovascular outcomes trial (CVOT) to prove it doesn’t cause excess heart risk. Before, companies typically ran blood-sugar trials and maybe smaller safety studies, but not massive multi-year heart safety trials. The science always said that a diabetes drug could have cardiovascular effects (positive or negative), yet the infrastructure to detect those effects (large, long-term trials in high-risk patients) wasn’t standard until Avandia’s situation essentially forced the FDA’s hand. Now, a decade later, those CVOTs have become the norm, enrolling thousands of patients for years to ensure no repeat of Avandia – a clear case of retrofitting accountability after the fact.
- JAK Inhibitors’ Delayed Warnings: Janus kinase (JAK) inhibitors (used for rheumatoid arthritis, ulcerative colitis, etc.) were hailed as game-changers and approved in the 2010s. Mechanistically, these drugs suppress parts of the immune system, so one could plausibly predict risks like serious infections, malignancies, or blood clots. Indeed, initial approvals came with some warnings, but it took years and a mandated post-market study to fully reveal the extent of these risks. In 2021, the FDA slapped class-wide boxed warnings on JAK inhibitors (tofacitinib, baricitinib, upadacitinib) after finding increased rates of heart attacks, strokes, cancers, and deaths in patients on tofacitinib. What’s notable is that two of those drugs (baricitinib and upadacitinib) had not yet shown those issues in trials – the FDA extrapolated the warning because they shared the same mechanism. In other words, they finally acted on the plausible class-wide risk proactively. But this was reactive policy born from hindsight; until then, companies were not required to have such large safety trials in high-risk populations prior to approval. The JAK example shows how even modern drug programs can “surprise” us with safety signals years post-approval – a sign that current pre-market testing wasn’t sufficient, and accountability for early signal detection was lacking.
- Biologics and Translational Gaps (the TGN1412 fiasco): Innovative therapies like monoclonal antibodies can have risks that animal models don’t predict. The case of TGN1412 in 2006 (often called the “Elephant Man trial” in the press) is a sobering example. This was a super-agonist antibody that, in theory, could treat autoimmune disease by stimulating a regulatory immune pathway. All the standard preclinical tests were done – including studies in monkeys that showed no serious issues – and regulators gave the go-ahead for a first-in-human trial. But when TGN1412 was infused into six healthy volunteers, it caused a catastrophic cytokine storm, sending all six into intensive care with multi-organ failure. The key revelation later was that the animal species used (cynomolgus monkeys) didn’t have the same immune response – the dangerous effect was specific to human immune biology. TGN1412 had “passed” the usual safety checklist, but the checklist wasn’t smart enough to account for a novel mechanism of action. Only after this near-disaster were new guidelines issued (in Europe) for first-in-human trials with high-risk biologics, including starting dose rules and better preclinical immune system assays. TGN1412 is now a canonical warning that our models can fail, and only rigorous caution and rapid response protocols can contain the damage when they do.
- Ranitidine and Hidden Impurities: Sometimes the risk is not from the drug’s intended effect at all, but from an impurity or a chemical instability that went unnoticed for decades. Ranitidine (Zantac), a popular heartburn drug since the 1980s, was considered very safe for a long time. However, improved analytical methods in recent years discovered that ranitidine can degrade to form NDMA, a potent carcinogen, especially under certain storage conditions. In 2019, regulators around the world found NDMA contamination in ranitidine products and moved to recall them. This was a shock – how could a 40-year-old drug suddenly become dangerous? In truth, low levels of NDMA may have been generated or present all along, but only modern high-sensitivity tests and focused scrutiny (sparked by independent laboratory findings) brought it to light. The “ranitidine affair” underscores that our analytical diagnostic layer is also evolving. Better tools can unexpectedly reveal that something we took for granted isn’t actually under control. When that happens, the system again must act swiftly – which the FDA did in eventually requesting all ranitidine be pulled from the U.S. market. It’s a reminder that vigilance can never ease up, even for legacy products. The thread through all these examples:
“Our diagnostic stack is still imperfect, and surprises will keep coming”
The only rational response is not to loosen scrutiny in hopes of speed, but to tighten accountability so that when a risk is plausible, it is aggressively investigated and addressed, not swept under the rug.
Founders and Investors: Ask the Right Questions
So, if you’re running or evaluating a biotech program, how do you ensure it’s “safe enough”? The counterintuitive answer: don’t just ask for more data; ask for the right questions and systems. Here are some practical takeaways.
For Founders (and R&D leaders): Instead of asking “Do we have enough data to feel safe?”, ask:
- What risk is plausible even if we haven’t seen it yet? Think mechanistically and theoretically. Anticipate your blind spots. (e.g., “Our drug affects enzyme X which is in the brain – could it cause neurological side effects down the road, even if animals didn’t show it?”)
- What assay or endpoint would detect that risk early? If you have a concern in mind, are you actively looking for it? Build a test or a measurement into your next study specifically to hunt for that issue.
- Who has the authority to stop or pause the program if a signal emerges? Identify that person or committee now, and make it clear they have executive support to call a time-out for safety. It might be your head of R&D, a safety review board, or even an external advisor – but define it clearly.
- What would regulators demand if they knew about this plausible risk? Don’t wait for the FDA to catch you off guard. If your drug’s mechanism suggests, say, cardiac risk, you can bet regulators (or advisory committees) will eventually zero in on that. Preempt it: run the study or collect the data that you know they would require if they were as worried as you are.
- Are we building just a drug submission, or a safety system? It’s easy to focus on the package you need to submit for approval – meeting the letter of each guideline. But also ask if you are building the internal systems (people, processes, decision triggers) that will make sure you truly know your drug’s safety profile. A culture of safety governance will outlast any single submission; it’s an asset that makes all your future programs more robust.
For board members (and Investors): When evaluating a company’s pipeline, look beyond the glossy slides. Probe their safety culture and plans:
- Is safety managed as a governance system or just a checklist? Does the team have routine safety review meetings, independent safety oversight, clear stop criteria? Or do they only discuss safety when a formal report is due? Governance mindset = good; checkbox mentality = risk.
- Active signal detection or passive hope? Ask if the company has any active pharmacovigilance plans before approval (like engaging external databases, early adopter patient monitoring, etc.). If all they have is “we’ll collect adverse event forms and review them,” that’s a red flag. Active systems catch problems in time; passive systems often don’t.
- Trial endpoints: designed for marketing or for safety? It’s common to see trials designed to maximize efficacy signals (which is fine) but check if they also include endpoints to detect predicted harms. For instance, a new immunotherapy might tout a surrogate efficacy endpoint, but did they include thorough immune-related toxicity monitoring or is it just hoping nothing bad happens? A trial that only looks at the positives might miss the negatives until it’s too late.
- Is toxicology (and safety expertise) embedded early? Did the company involve experienced toxicologists and safety physicians in candidate selection and early development, or are they scrambling post-Phase II when an issue popped up? Early engagement can prevent investing in a fundamentally unsafe compound. Late engagement means a nasty surprise might be around the corner.
One more tip: remember that a program’s valuation usually doesn’t collapse merely because a risk exists; it collapses when it becomes clear that the team failed to control that risk. In other words, markets can tolerate drugs with risks (many drugs have serious side effects and yet succeed commercially), but they punish companies that appear negligent or clueless about those risks. When you do your due diligence, a solid safety infrastructure is actually a protector of value.
Bottom Line
Thalidomide and Vioxx were not “unpredictable, unfortunate one-offs” – they were system failures that allowed known or knowable dangers to fester until disaster struck. In both cases, and in others since, the root cause was that safety insights existed but were not empowered. The only thing that forced action was public accountability after people were hurt.Safety isn’t an advisory function; it’s a system. And like any system, it only works when someone is truly responsible for its outcomes. If your safety strategy depends on “good people eventually doing the right thing,” it is brittle and likely to fail under pressure. The only durable approach is to bake in a structure that ties safety signals to real authority and rapid action. That means giving teams the tools to see problems and the mandate to act on them without hesitation. This is the essence of strong regulatory toxicology and pharmacovigilance – not paperwork, but governance of risk
Data is just noise without the authority to act
In the end, drug development will always involve uncertainty and the potential for harm. Embracing accountability in the face of that uncertainty isn’t just the ethical choice; it’s the pragmatic one. It prevents tragedies, protects patients, and ultimately differentiates the companies that can be trusted with advancing medicine. Safety done right may not grab headlines, but it’s the foundation of every truly successful therapy. After all, a breakthrough drug is only a breakthrough if it’s safe enough for people to benefit from it – and ensuring that is a responsibility that cannot be abdicated.
References
- Vargesson, N. (2015). Thalidomide-induced teratogenesis: History and mechanisms. Birth Defects Research Part C: Embryo Today: Reviews, 105(2), 140–156. https://doi.org/10.1002/bdrc.21096. https://pubmed.ncbi.nlm.nih.gov/21507989/
- Nau, H., Scott, W. J., & Shepard, T. H. (2022). Comparison of rat and rabbit embryo–fetal developmental toxicity data for 379 pharmaceuticals: On the nature and severity of developmental effects. Birth Defects Research, 114(4), 191–212. https://doi.org/10.1002/bdr2.2006. https://pmc.ncbi.nlm.nih.gov/articles/PMC8865449/
- McGettigan, P., & Henry, D. (2006). What have we learnt from Vioxx? BMJ, 332(7553), 130–133. https://doi.org/10.1136/bmj.332.7553.130. https://pmc.ncbi.nlm.nih.gov/articles/PMC1779871/
- Topol, E. J. (2004). Failing the public health—Rofecoxib, Merck, and the FDA. New England Journal of Medicine, 351(17), 1707–1709. https://doi.org/10.1056/NEJMp048286
- Institute of Medicine (US) Committee on the Assessment of the US Drug Safety System. (2007). The future of drug safety: Promoting and protecting the health of the public. National Academies Press. https://doi.org/10.17226/11750 https://www.ncbi.nlm.nih.gov/books/NBK52924/
- Psaty, B. M., & Furberg, C. D. (2005). COX-2 inhibitors—Lessons in drug safety. New England Journal of Medicine, 352(11), 1133–1135. https://doi.org/10.1056/NEJMsb050033
- U.S. Food and Drug Administration. (2016). More than 7 years of hindsight: Revisiting the FDA’s 2008 guidance on cardiovascular outcomes trials for type 2 diabetes medications. Therapeutic Innovation & Regulatory Science, 50(6), 711–718. https://doi.org/10.1177/2168479016664234. https://pmc.ncbi.nlm.nih.gov/articles/PMC5070965/
- U.S. Food and Drug Administration. (2021, September 1). FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions. https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death
- Suntharalingam, G., Perry, M. R., Ward, S., Brett, S. J., Castello-Cortes, A., Brunner, M. D., & Panoskaltsis, N. (2006). Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. New England Journal of Medicine, 355(10), 1018–1028. https://doi.org/10.1056/NEJMoa063842
- Stebbings, R., Findlay, L., Edwards, C., Eastwood, D., Bird, C., North, D., … Thorpe, R. (2009). Safety of biologics, lessons learnt from TGN1412. Current Opinion in Biotechnology, 20(6), 673–677. https://doi.org/10.1016/j.copbio.2009.10.002 https://www.sciencedirect.com/science/article/abs/pii/S095816690900130X
- Zhang, Y., et al. (2022). N-nitrosodimethylamine (NDMA) contamination of ranitidine products: A review of recent findings. Pharmaceuticals, 15(7), 844. https://doi.org/10.3390/ph15070844. https://pmc.ncbi.nlm.nih.gov/articles/PMC9261846/
- Thermo Fisher Scientific. (n.d.). Analysis of nitrosamine impurities in ranitidine. https://www.thermofisher.com/sg/en/home/global/forms/industrial/ranitidine.html