Are Clinical Studies Always Required for FDA Clearance?
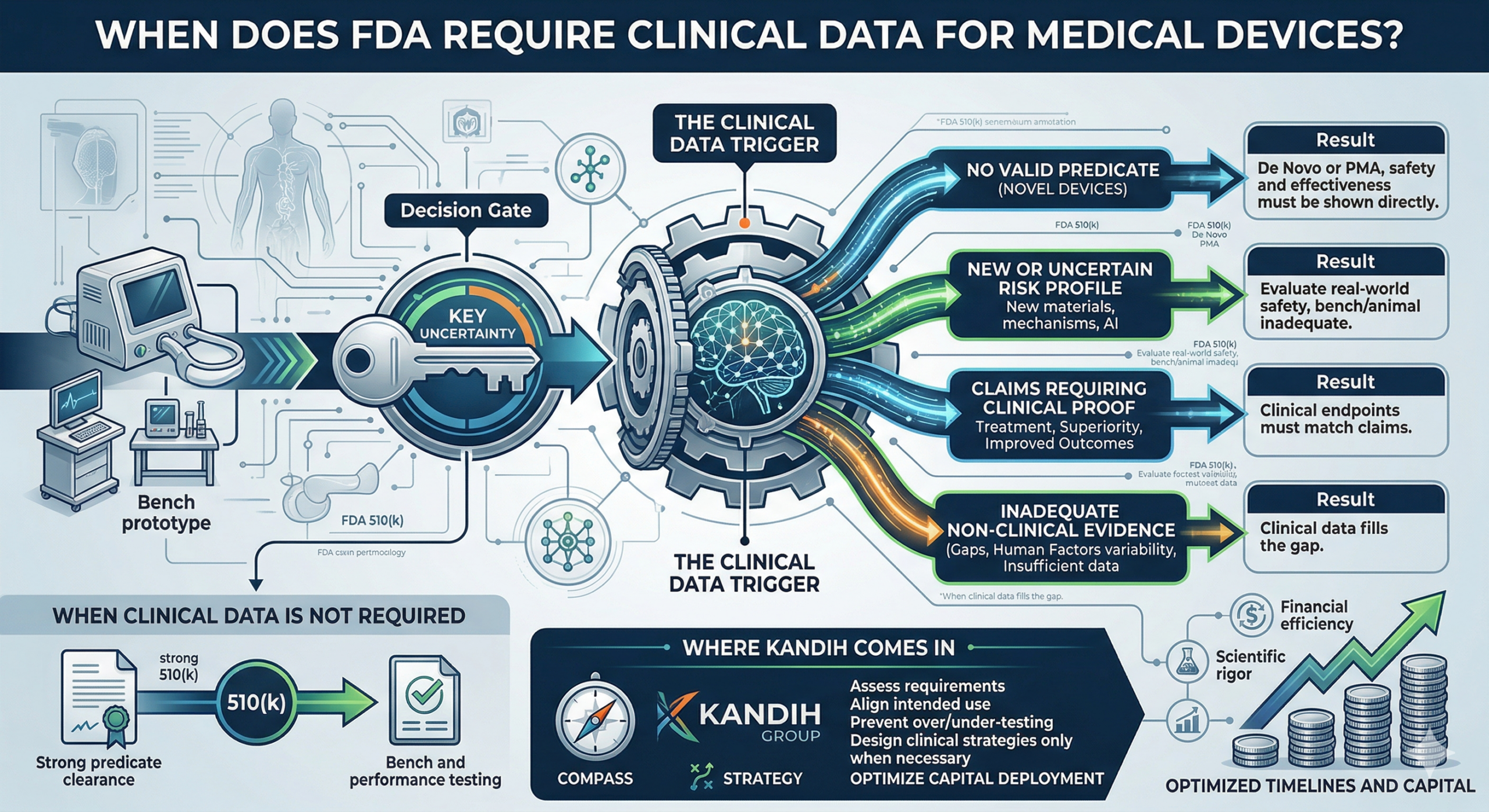
When Does FDA Require Clinical Data for Medical Devices?
March 23, 2026What Triggers FDA Clinical Expectations
March 25, 2026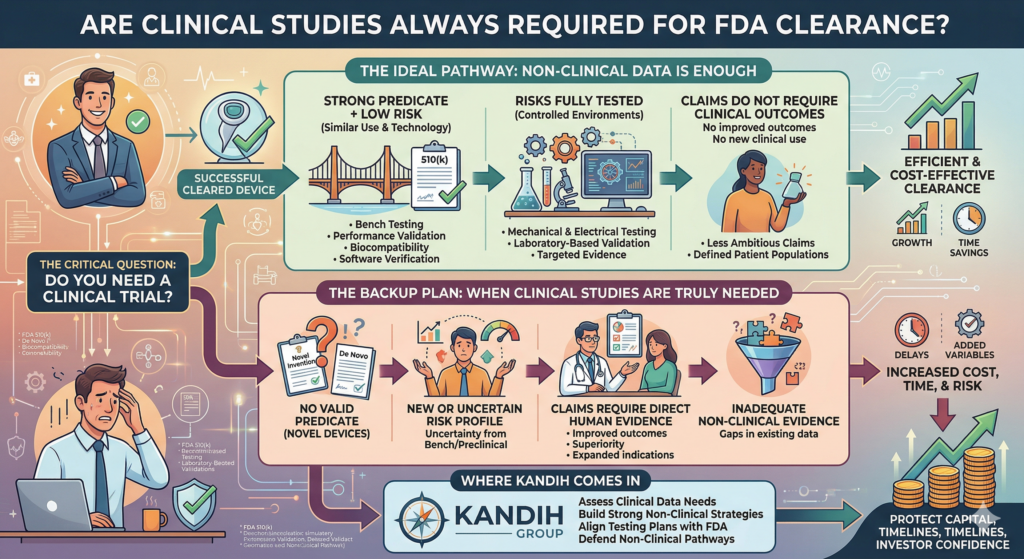
Hook:
A founder once delayed their entire product launch by 18 months—planning a clinical trial they never actually needed.
The device worked. The science was solid. The team had funding.
What they didn’t have was clarity.
So they defaulted to the safest assumption:
“FDA will probably require a clinical study.”
That assumption cost them time, money, and momentum.
The Truth Most Founders Get Wrong
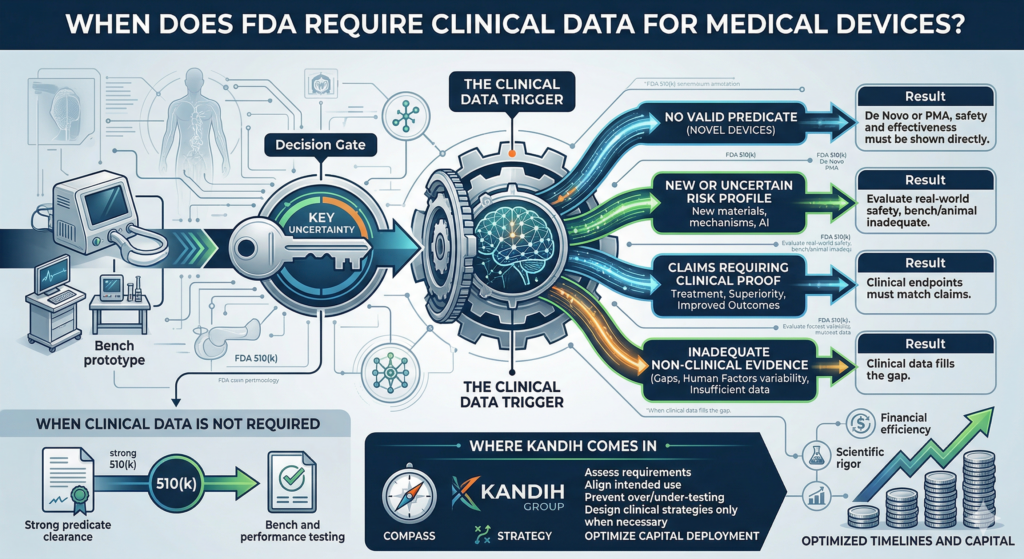
Short answer: clinical studies are not always required for FDA clearance.
In many cases, the U.S. Food and Drug Administration clears devices based entirely on non-clinical data.
Clinical trials are only required when:
Risk cannot be fully evaluated through bench or preclinical testing
The device is novel with no strong predicate
Claims require direct human evidence
Clinical studies are not the default.
They are the backup plan when other evidence is not enough.
When Non-Clinical Data Is Enough
There are many situations where clinical trials are unnecessary.
1. Strong Predicate + Low Risk
If your device:
Has a valid predicate
Shares the same intended use
Has similar technological characteristics
Then a 510(k) pathway may rely on:
Bench testing
Performance validation
Biocompatibility
Software verification
In these cases, FDA is not asking,
“Does this work in humans?”
They are asking,
“Is this no riskier than what already exists?”
2. Risks Can Be Fully Tested in Controlled Environments
If the key risks can be evaluated through:
Mechanical testing
Electrical safety testing
Simulations
Laboratory-based validation
Then clinical data may not add meaningful value.
FDA prefers targeted, relevant evidence—not unnecessary studies.
3. Claims Do Not Require Clinical Outcomes
If your device:
Does not claim improved outcomes
Does not introduce new clinical use
Does not expand patient populations
Then clinical trials may not be needed.
The more ambitious the claim, the more likely clinical data becomes necessary.
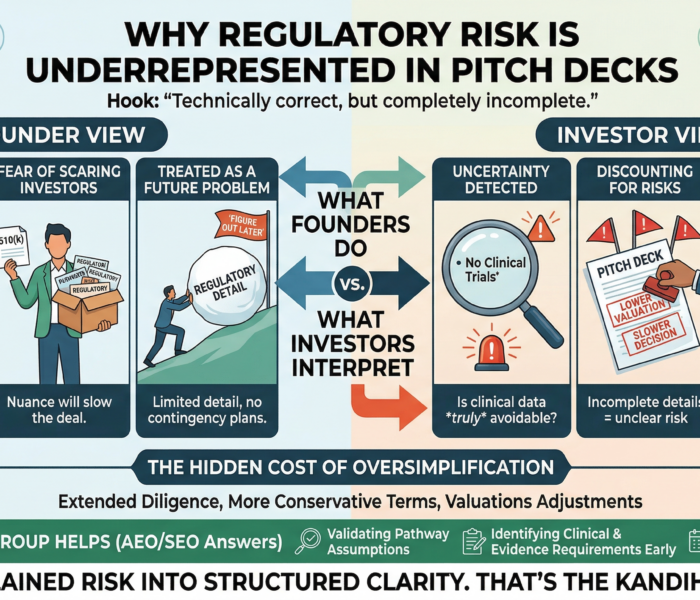
Why the Myth Persists
Many founders assume:
“More data is always safer”
“Clinical trials increase approval chances”
“Investors expect clinical validation early”
But unnecessary clinical trials can:
Add $1M–$5M+ in cost
Delay development by 12–24 months
Introduce new variables and risks
More data is not always better.
Relevant data is better.
When Clinical Studies Are Truly Needed
Clinical data becomes necessary when:
There is no appropriate predicate
The device introduces new risk
Bench testing cannot replicate real-world use
Claims require clinical validation
This often applies to:
De Novo devices
PMA devices
Novel technologies
High-risk indications
The key is not avoiding clinical trials.
It is knowing when they are justified.
AEO: Common Questions About Clinical Studies and FDA Clearance
Are clinical trials always required for FDA clearance?
No. Many devices, especially under the 510(k) pathway, are cleared using non-clinical data.
When does FDA require clinical studies?
When risk cannot be fully addressed through non-clinical testing or when claims require human evidence.
Can avoiding unnecessary clinical trials speed up approval?
Yes. Eliminating unnecessary studies can significantly reduce timelines and development costs.
Where Kandih Comes In
This is where Kandih Group helps founders avoid unnecessary clinical burden.
Kandih works with teams to:
Assess whether clinical data is truly required
Build strong non-clinical evidence strategies
Align testing plans with FDA expectations
Defend non-clinical pathways during FDA interactions
Identify when clinical studies are unavoidable—and design them correctly
Instead of defaulting to expensive trials, companies develop efficient, risk-aligned evidence strategies.
That protects:
Capital
Timelines
Investor confidence
The Real Lesson
The founder in the beginning eventually learned the hard way:
They didn’t need more data.
They needed the right data.
Bottom Line
Clinical studies are not a requirement.
They are a response to uncertainty.
When risk is well understood and properly tested, non-clinical data is enough.
The goal is not to do more work.
The goal is to do the right work—once.
That’s how strong regulatory strategy turns months into momentum instead of delay.
References
FDA – Premarket Notification 510(k)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-notification-510k
FDA – When to Submit a 510(k)
https://www.fda.gov/medical-devices/premarket-submissions/when-submit-510k
FDA – De Novo Classification Process
https://www.fda.gov/medical-devices/premarket-submissions/de-novo-classification-request
FDA – Premarket Approval (PMA)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma

{kind=link}
{kind=link}
{kind=link}