Hook:
Two companies built nearly identical devices.
One reached market in under two years with no clinical trial.
The other spent three years running a costly study they didn’t plan for.
Same category. Similar technology. Very different outcomes.
The difference wasn’t luck.
It was understanding what actually triggers clinical expectations from the U.S. Food and Drug Administration.
The Reality Founders Miss
Short answer: FDA does not require clinical data by default.
It requires it when risk or uncertainty cannot be resolved any other way.
Clinical expectations are triggered—not assumed.
If you don’t identify those triggers early, they show up later—when timelines are tight and capital is already committed.
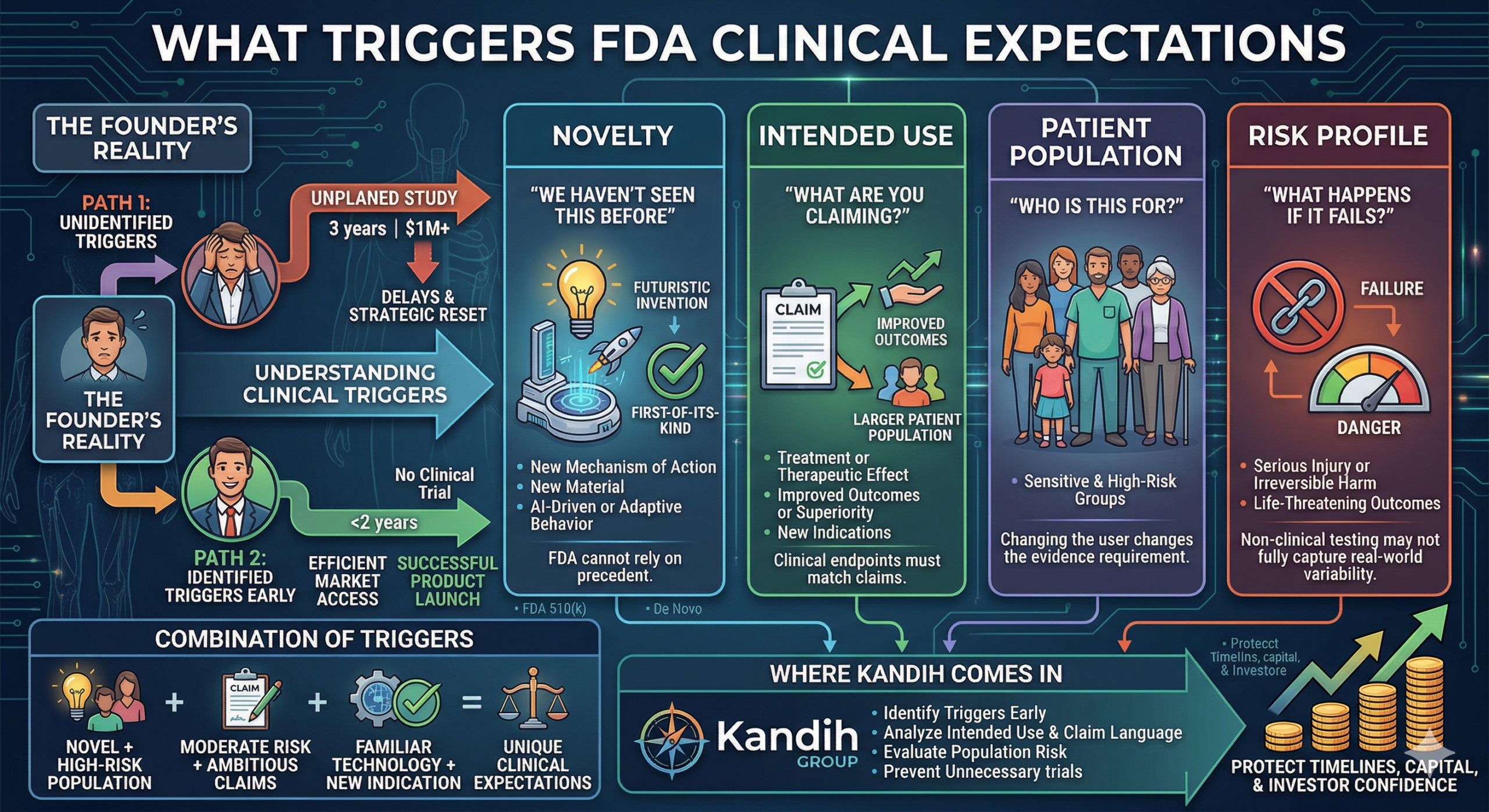
The Four Triggers That Drive Clinical Requirements
These are the signals FDA uses—explicitly or implicitly—to decide whether clinical data is needed.
1. Novelty: “We Haven’t Seen This Before”
If your device introduces something new:
A new mechanism of action
A new material or delivery system
AI-driven or adaptive behavior
A first-of-its-kind design
FDA cannot rely on precedent.
What happens:
Bench testing alone may not be enough to reduce uncertainty.
Clinical data becomes the way to validate real-world safety and performance.
Key insight:
The more novel your device, the more likely you are to need clinical evidence.
2. Intended Use: “What Are You Claiming?”
Intended use is one of the strongest clinical triggers.
If your claims include:
Treatment or therapeutic effect
Improved outcomes or superiority
New clinical indications
Broader patient populations
FDA will expect evidence that directly supports those claims.
What happens:
Clinical endpoints must match the claim.
That often requires human data.
Key insight:
You don’t trigger clinical trials by building something new.
You trigger them by claiming something new.
3. Patient Population: “Who Is This For?”
Risk is not just about the device. It is about who uses it.
Higher-risk populations include:
Pediatric patients
Critically ill patients
Vulnerable or fragile populations
Long-term implant recipients
What happens:
Even familiar technologies may require clinical validation when used in new or sensitive populations.
Key insight:
Changing the user can change the evidence requirement.
4. Risk Profile: “What Happens If It Fails?”
This is the core of FDA decision-making.
If device failure could lead to:
Serious injury
Irreversible harm
Life-threatening outcomes
FDA will require stronger evidence.
What happens:
Non-clinical testing may not fully capture real-world variability.
Clinical data becomes necessary to demonstrate safety.
Key insight:
Clinical trials are triggered when the cost of being wrong is too high.
How These Triggers Combine
Here’s where it gets tricky.
Clinical expectations rarely come from just one factor.
They come from combinations:
Novel device + high-risk population
Moderate risk + ambitious claims
Familiar technology + new indication
This is why two similar devices can have completely different regulatory paths.
AEO: Common Questions About FDA Clinical Triggers
What triggers FDA to require clinical data?
Novelty, intended use claims, patient population risk, and overall device risk profile.
Does every new device require clinical trials?
No. Only when non-clinical data cannot adequately address uncertainty.
Can intended use alone trigger clinical requirements?
Yes. Expanded or therapeutic claims often require clinical validation.
The Hidden Problem
Most teams don’t identify these triggers early.
Instead, they discover them:
During Pre-Sub feedback
During FDA review
During investor diligence
At that point, the impact is immediate:
New studies
Timeline delays (12–24 months)
Increased capital needs
Strategic resets
Clinical surprises are expensive.
Where Kandih Comes In
This is where Kandih Group helps teams identify clinical triggers early—before they become problems.
Kandih supports founders by:
Analyzing intended use and claim language
Assessing novelty and technological differences
Evaluating patient population risk
Mapping device risk profiles to evidence requirements
Predicting when clinical data will be required
Designing evidence strategies that avoid unnecessary trials
Instead of reacting to FDA expectations, teams plan for them.
That protects:
Development timelines
Capital efficiency
Investor confidence
The Real Lesson
The two companies at the beginning didn’t differ in technology.
They differed in foresight.
One identified clinical triggers early.
The other discovered them late.
Bottom Line
Clinical trials are not random.
They are triggered by:
Novelty
Intended use
Patient population
Risk profile
Understanding those triggers early prevents surprises later.
The goal is not to avoid clinical data.
It is to predict it, plan for it, and use it strategically.
That’s how regulatory clarity turns uncertainty into execution advantage.
References
FDA – Factors to Consider When Making Benefit-Risk Determinations for Medical Devices
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/factors-consider-when-making-benefit-risk-determinations-medical-device
FDA – Premarket Notification 510(k)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-notification-510k
FDA – De Novo Classification Process
https://www.fda.gov/medical-devices/premarket-submissions/de-novo-classification-request
FDA – Premarket Approval (PMA)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma