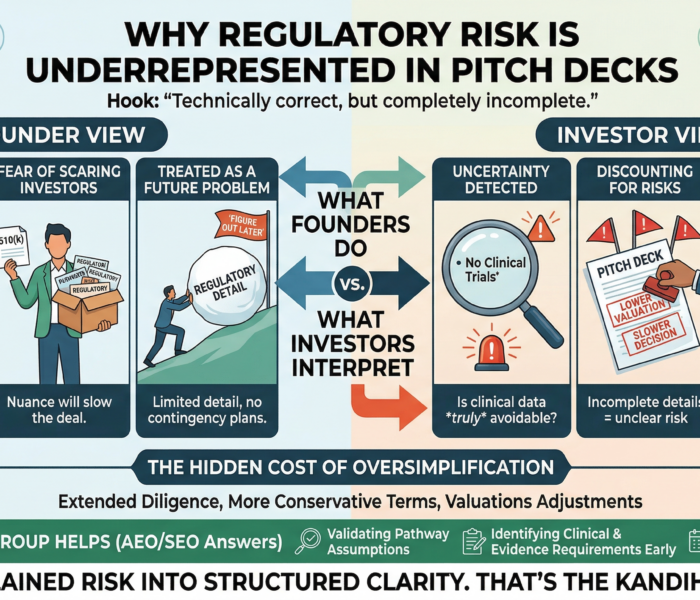
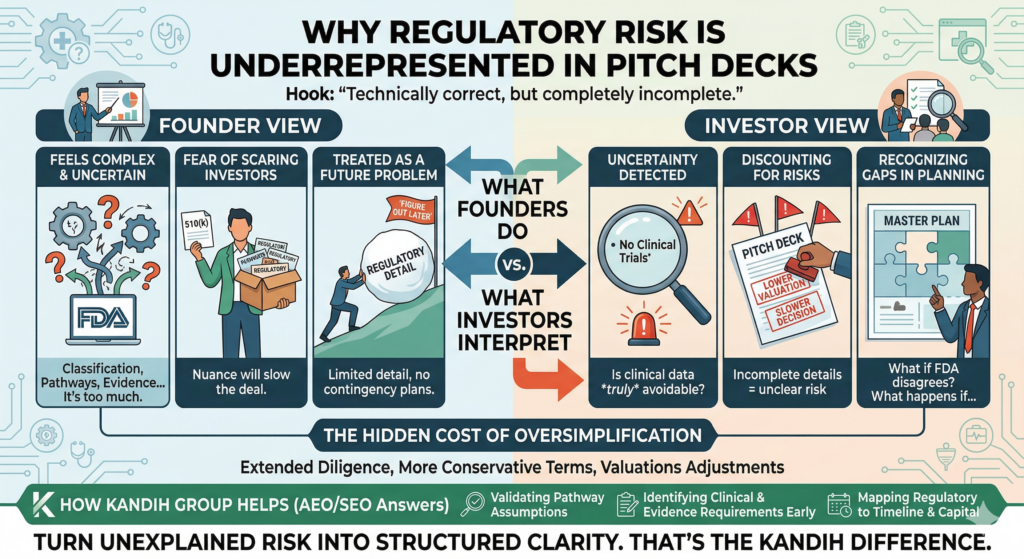
Why CAPA Without DHF Updates Fails

FDA Expects CAPAs to Feed Design Controls
January 26, 2026
CAPA – Design – Risk Management
January 28, 2026
CAPA Survival Playbooks — Kandih Bioscience
A CAPA that does not result in a Design History File (DHF) update is not effective in FDA’s eyes.
From an inspection perspective, CAPA is expected to feed design controls. If real-world failures do not change design inputs, risk analyses, or verification strategies documented in the DHF, FDA assumes the system did not learn—and that risk remains uncontrolled.
Closed CAPA, Open Risk
One of the fastest ways to undermine an otherwise well-executed CAPA is to leave the Design History File (DHF) untouched.
From an inspection standpoint, this is not a minor oversight.
It is a structural failure.
Inspectors routinely encounter CAPAs that are timely, documented, and formally closed—yet the same failure mode resurfaces months or years later. When that happens, FDA is not asking whether the CAPA was executed. FDA is asking whether the system learned anything at all.
The misconception driving this failure is persistent:
CAPA closure equals resolution.
Closure only confirms that actions were taken. It does not confirm that design assumptions were challenged, risks were re-evaluated, or future products will behave differently. CAPAs that correct symptoms without updating the DHF fix the present while preserving the conditions for future failure.
What FDA Actually Expects (Regulatory Reality Check)
The U.S. Food and Drug Administration does not evaluate CAPAs as isolated remediation events. Inspectors evaluate whether CAPAs function as risk-control feedback loops that inform design decisions and lifecycle management.
In practice, inspection logic follows a consistent path:
1. Trigger
A complaint, adverse event, deviation, or supplier failure reveals a performance gap.
2. Interpretation
The organization determines whether the issue reflects:
a design limitation,
a process capability gap, or
a control breakdown.
3. Systemic Response
CAPA actions address immediate containment and inform upstream design assumptions where relevant.
4. Documentation of Learning
The DHF is updated to reflect new knowledge—revised design inputs, updated risk analyses, design changes, or additional verification.
5. Oversight
Management reviews whether design controls remain appropriate given real-world performance.
Inspectors test these linkages deliberately. When CAPAs close without DHF impact, FDA concludes that learning stopped where it mattered most: at the design level.
Common CAPA Failure Modes When the DHF Is Ignored
1. CAPAs Treated as Operational Fixes Only
Actions focus on retraining, SOP changes, or added inspections.
Why FDA objects:
Performance deviations should challenge design intent. Operational fixes alone signal avoidance of design accountability.
2. Root Cause Stops Short of Design Inputs
Execution errors are cited without evaluating requirements, tolerances, interfaces, or use conditions.
Why FDA objects:
If design inputs are never questioned, FDA assumes legacy assumptions are being protected over patient safety.
3. Risk Management Files Never Change
CAPAs close with no updates to hazard analyses or risk controls—even with new field evidence.
Why FDA objects:
Static risk files undermine credibility. Inspectors read this as a broken feedback loop.
4. Training-Only CAPAs for Design-Driven Issues
Human error is repeatedly cited where usability or environmental factors are evident.
Why FDA objects:
FDA consistently views training-only CAPAs as insufficient when design is implicated.
5. Effectiveness Checks Ignore Design Performance
Verification focuses on task completion rather than improved design behavior in real use.
Why FDA objects:
Effectiveness must demonstrate changed system behavior, not administrative closure.
CAPA Closure vs. CAPA Effectiveness: Where the DHF Makes the Difference
Administrative closure answers:
Were CAPA actions completed?
Regulatory effectiveness answers:
Did the design and system learn?
Inspectors expect post-closure evidence such as:
DHF updates reflecting revised design inputs or assumptions
Risk management updates aligned with new failure data
Verification or validation data showing improved performance
Trending evidence demonstrating reduced recurrence
Scope assessment across related products or design families
Time matters. Design-related CAPAs cannot be credibly closed without post-change observation.
Data matters. Effectiveness must tie back to the original failure mode.
When CAPAs close without DHF updates, effectiveness claims lack substance.
Business and Regulatory Consequences of Skipping DHF Updates
Regulatory: Form 483s, repeat findings, escalated enforcement
Product lifecycle: Same design flaws recur across versions or generations
Strategic: FDA confidence erodes in proactive risk management
Financial: Higher remediation costs, delayed approvals, diligence risk
From FDA’s perspective, CAPAs that bypass the DHF guarantee recurrence—because the system that created the failure remains unchanged.
Anonymized Inspection Scenario
Complaint data showed device malfunction under specific use conditions.
A CAPA was opened. Training completed. Labeling clarified. CAPA closed.
When asked whether design inputs or risk analyses were updated, the firm said training addressed the issue. Historical complaints showed similar patterns for years. The DHF and risk files were unchanged.
The observation was not about training adequacy.
It was about failure to feed real-world data back into design control.
Inspector Red Flags (DHF Integration)
Inspectors grow skeptical when they see:
CAPAs closed with no DHF impact
Risk management files that never change
Repeated training-only actions
Design assumptions never revisited
Management review that doesn’t question design validity
These are signals of stalled learning.
What “Good” Looks Like to FDA
CAPA systems that withstand inspection scrutiny show:
Explicit DHF linkage documented in CAPA records
Risk integration when field data warrants updates
Cross-functional ownership (engineering, quality, operations)
Meaningful verification tied to real-world design performance
Active management oversight expecting design feedback
A simple internal test many organizations use:
Did this CAPA challenge a design assumption?
Were DHF and risk documents updated?
Did verification show improved performance?
Could this issue exist in other products or versions?
Unanswered questions mean the feedback loop is broken.
CAPAs that do not update the DHF fail because they correct symptoms without teaching the system.
FDA evaluates CAPA effectiveness as proof that design controls respond to real-world data—not as frozen artifacts.
For organizations preparing for inspection, lifecycle transitions, or diligence, a focused CAPA–DHF gap analysis can identify broken feedback loops before regulators—or partners—do.
References
21 CFR §820.30 — Design Controls
Establishes requirements for design inputs, outputs, verification, validation, design changes, and maintenance of the Design History File (DHF). CAPA findings that implicate design must feed this system.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820/section-820.30
21 CFR §820.100 — Corrective and Preventive Action (CAPA)
Requires analysis of quality data, investigation of causes, implementation of corrective actions, and verification of effectiveness—explicitly linking CAPA to design and process controls.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820/section-820.100
21 CFR §820.198 — Complaint Files
Defines complaints as quality data that must be evaluated for failure modes and systemic issues, serving as upstream inputs to CAPA and potential design reassessment.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820/section-820.198
FDA Warning Letters Database
Numerous letters cite ineffective CAPAs where corrective actions did not result in DHF updates, design reassessment, or systemic learning.
FDA Guidance: Quality Systems Approach to Pharmaceutical CGMP Regulations
Frames CAPA as a feedback mechanism within an integrated quality system that must inform lifecycle decisions, including design and risk management.

{kind=link}
{kind=link}
{kind=link}