When to File a New 510(k): Post-Change Decision Framework

CGM Sensors and Hidden Plasticizers: Reddit’s CGM Material Fears
June 22, 2026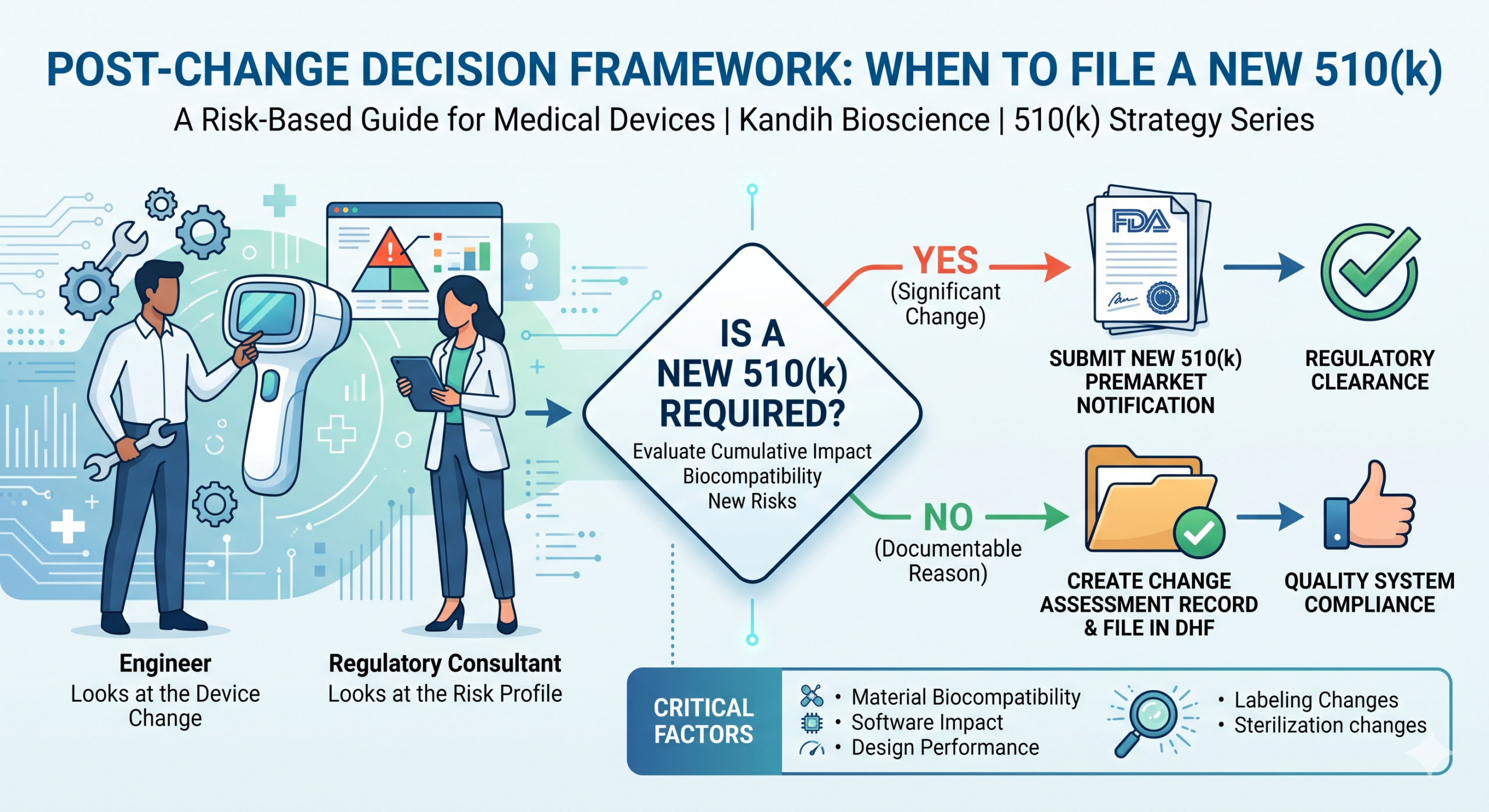
By Kandih Bioscience • 510(k) Strategy Series
From r/FDA510kSupport: “We just changed our device housing material. Same function, cleaner aesthetics, similar polymer family. Our engineer says it’s a minor change. Our regulatory consultant went quiet for three days and came back saying we might need a new 510(k). Who is right and how do we even figure this out?”
Both of them might be right. That’s the uncomfortable truth about post-market device changes.
The engineer is looking at the change. The regulatory consultant is looking at the risk profile of that change. They’re not actually disagreeing — they’re answering two different questions. And until you answer the right one, you can’t know whether you need a new 510(k) or not.
This is one of the most common and most costly miscalls in post-market device management. A company makes a change that genuinely seems minor, skips the formal change assessment, ships the updated device, and finds out 18 months later — during an FDA inspection, or after a competitor complaint, or during due diligence for a Series B — that the change required a new submission they never filed.
This piece walks through how FDA actually evaluates post-market changes, where the decision framework breaks down, and what a sound change assessment process looks like in practice.
Why This Is Harder Than It Sounds
FDA doesn’t give you a simple list that says: ‘these changes need a new 510(k), these don’t.’ What it gives you instead is a risk-based framework — a set of questions you’re supposed to work through for every change, then document your reasoning.
The primary guidance document is FDA’s 2017 guidance, “Deciding When to Submit a 510(k) for a Change to an Existing Device.” It covers changes to design, materials, software, labeling, manufacturing, and performance specifications. And the honest read of that document is: it depends.
It depends on whether the change could affect safety or effectiveness. It depends on whether the change introduces a new risk. It depends on whether the original 510(k) data still covers the modified device. It depends on how your device is classified and what the original predicate relationship looked like.
None of those are yes or no questions. All of them require documented reasoning. And the consequence of getting it wrong — in either direction — is real.
Filing a new 510(k) when you didn’t need to costs time and money. Not filing when you should have costs your 510(k) clearance. The second mistake is the one that creates legal liability, triggers warning letters, and kills acquisition conversations mid-process.
The Framework FDA Expects You to Use
FDA’s change assessment framework asks you to evaluate every device change through two lenses:
- Could this change affect the safety or effectiveness of the device?
- Could this change affect the device’s substantial equivalence to its predicate?
If the answer to either is yes — or could reasonably be yes — a new 510(k) is likely required. If you can document a clear ‘no’ to both, you may be able to proceed without one. But that ‘no’ has to be written down, signed, and kept in your design history file. It cannot live only in someone’s memory.
Across the specific change types FDA’s guidance addresses, here’s how the framework plays out:
Change Type → What Triggers a New 510(k)
Material change → New 510(k) if the material contacts the patient and original biocompatibility data doesn’t cover the new material
Design change → New 510(k) if it changes how the device performs its intended function or introduces new failure modes
Software change → New 510(k) if the change affects device safety or the device is Software as a Medical Device (SaMD)
Labeling change → New 510(k) if it expands indications, changes intended use, or introduces new patient populations
Manufacturing change → New 510(k) if it could affect device performance, sterility, or biocompatibility
Sterilization change → New 510(k) almost always — sterilization affects safety and biocompatibility directly
These aren’t mechanical rules. They’re starting points for a documented risk analysis. The mistake most companies make is treating them as final answers.
The Three Places the Decision Goes Wrong
After working through post-market change assessments with device companies, the same failure patterns show up. Not because companies are careless. Because the framework gives you room to rationalize in the wrong direction.
Failure Point #1: Evaluating the change in isolation.
A material change that looks minor by itself can tip into a new 510(k) requirement when you account for what else changed recently. If your manufacturing process also shifted in the last two years, and your sterilization validation was updated, and now the material is changing too — those three changes together may cross a threshold that none of them would have crossed individually.
FDA specifically looks at the cumulative impact of changes since the last clearance. Each change should be assessed not just on its own merits but in the context of what else has changed.
Failure Point #2: Confusing material similarity with material equivalence.
This is exactly what the Reddit post at the top of this piece describes. A polymer that’s in the same family as the original material is not automatically biocompatibility-equivalent to the original. Different formulations can have different additive packages, different colorants, different surface chemistry, different extractables profiles.
If the new material hasn’t been characterized under ISO 10993-18 and your existing biocompatibility data was generated on the original material, the data gap is real — regardless of how similar the materials seem on spec sheets.
Failure Point #3: Treating the change assessment as a team conversation instead of a document.
A lot of change assessments happen in meetings. Engineers, regulatory people, and sometimes outside consultants sit around a table, agree the change is minor, and move on. No written analysis. No documented risk reasoning. No sign-off tied to a specific version of the device.
Two years later, nobody can reconstruct the decision. The FDA inspector who shows up during an audit isn’t asking for your meeting notes. They’re asking for your change assessment record. If it doesn’t exist, the inspection finding is automatic.
For investors: if a device company you’re evaluating has made post-market changes — materials, design, software, labeling — ask specifically whether those changes were assessed under FDA’s 510(k) change guidance and whether the assessments are documented in the design history file. A ‘yes’ with documentation is a green flag. A ‘we discussed it and decided it was fine’ without a paper trail is a regulatory liability sitting in the quality system.
What a Sound Change Assessment Process Actually Looks Like
A change assessment isn’t a bureaucratic exercise. It’s a structured way of answering a specific question: does the change we made require us to go back to FDA before we ship the updated device?
A credible change assessment has four components:
- A clear description of the change — what exactly changed, at which version, on which date, compared to which prior configuration
- An impact analysis — does this change affect the device’s safety, effectiveness, or substantial equivalence to its predicate?
- A risk-based conclusion — based on FDA’s guidance, does this change require a new 510(k), a Special 510(k), or no new submission?
- Supporting evidence — if you’re concluding no new submission is required, what data or documentation supports that conclusion?
That process needs to happen before the change ships. Not after. Not during the next quality audit. Before the device with the change leaves the building.
For material changes specifically, the supporting evidence piece often includes biocompatibility data — either new testing on the changed material, or a written justification (with data) for why the existing biocompatibility record still applies. That’s not optional language. It’s what FDA reviewers look for when they evaluate a change assessment package.
Founders: the cost of a proactive change assessment is hours of documentation. The cost of retroactively reconstructing one under FDA scrutiny is weeks of work, often with a regulatory attorney involved, and sometimes with a voluntary recall or corrective action attached. Do it before you ship.
What This Means for Your Commercial Timeline
Post-market change decisions don’t just affect regulatory risk. They directly affect your ability to raise, sell, and scale.
Here’s what happens when a change assessment gap surfaces late:
- Acquisition due diligence: a buyer’s regulatory counsel reviews the change log and finds undocumented changes. The acquisition pauses for a compliance assessment. The conversation about price shifts.
- Series B or C diligence: an investor’s technical advisors flag that the device being sold isn’t exactly the device that was cleared. The financing round gets complicated until a regulatory response is filed.
- FDA inspection: an inspector requests the change assessment record for a material substitution. The record doesn’t exist. The Form 483 observation leads to a warning letter. Commercial activity stops while a CAPA is opened.
None of those scenarios start with a catastrophic failure. They all start with a change that seemed minor, wasn’t formally assessed, and wasn’t documented. The gap is small. The consequence, when it surfaces, is not.
Where Kandih Bioscience Comes In
The question of whether a change requires a new 510(k) is not always obvious. It depends on the specific change, the device’s classification, the original predicate relationship, and the cumulative history of changes since the last clearance.
At Kandih Bioscience, we help device companies work through exactly this kind of decision — before it becomes a compliance problem. That means:
- Reviewing your change log and assessing each change against FDA’s 510(k) change guidance to identify which changes were correctly handled and which ones may have a documentation gap
- Building a change control process your team can run consistently — so every future change gets assessed, documented, and concluded before it ships
- Evaluating material changes specifically for biocompatibility impact — including whether your existing ISO 10993 data still applies to the modified device or whether new testing is needed
- Preparing change assessment packages that are defensible under FDA inspection, investor due diligence, and acquisition review
If you’ve made post-market changes to a cleared device and you’re not confident the documentation is solid, the right time to review it is now — not when an inspector asks or a deal is on the table.
→ Book a Biocompatibility Review with Kandih: kandih.com/bio_compatibility
Contact Kandih Bioscience • info@kandih.com • kandih.com • 240.565.8933
References
1. FDA — Deciding When to Submit a 510(k) for a Change to an Existing Device (2017)
2. FDA — Premarket Notification 510(k): Overview and Guidance (current)
3. FDA — Use of International Standard ISO 10993-1: Biological Evaluation of Medical Devices (2020)
5. FDA — Quality System Regulation: Design Controls and Change Management (21 CFR Part 820) (current)
6. FDA — Special 510(k) Program Guidance (2019)