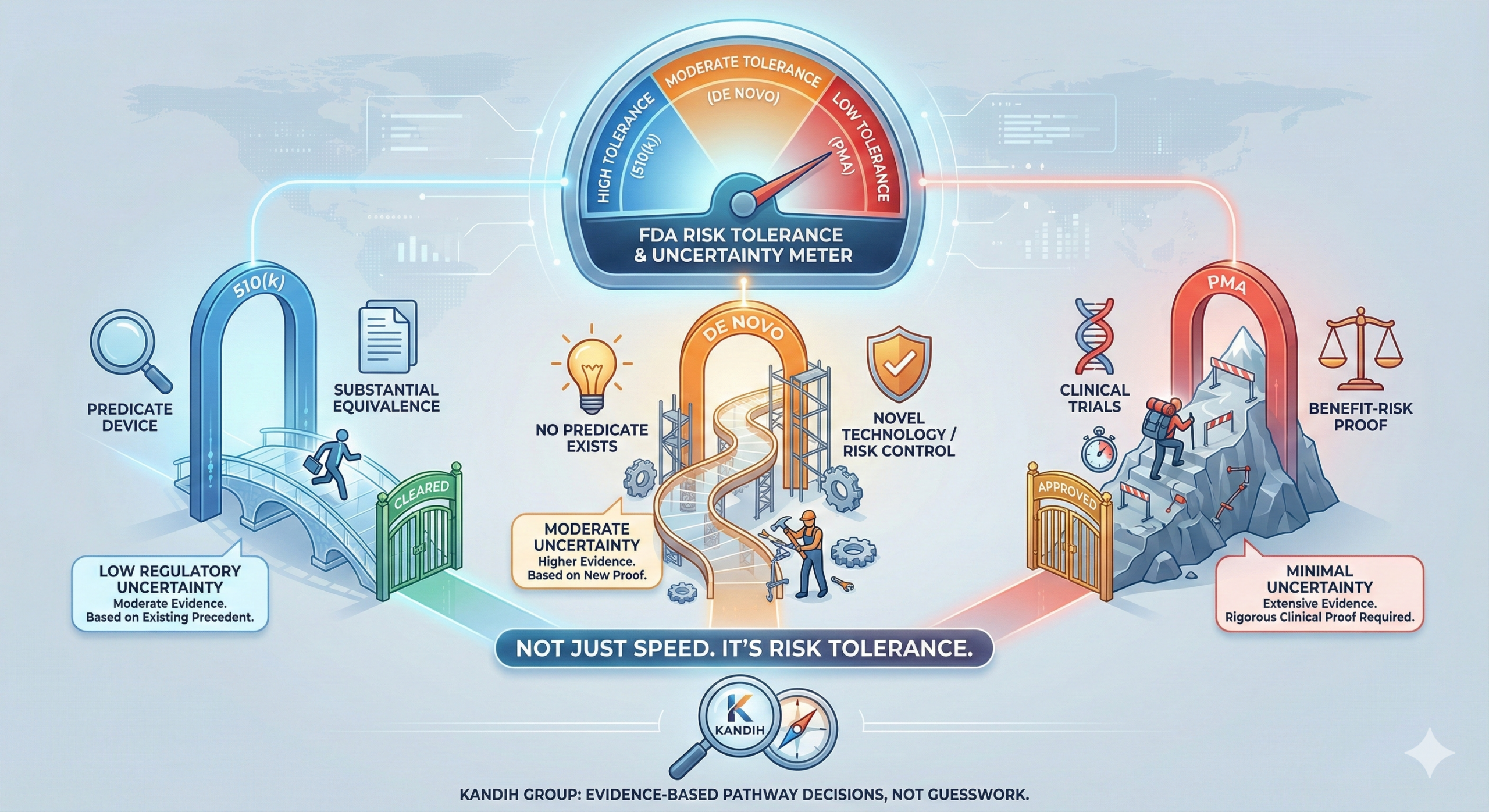
Short answer: the real difference between 510(k), De Novo, and PMA is not speed. It is risk tolerance.
Each pathway reflects how much uncertainty the U.S. Food and Drug Administration is willing to accept—and how much evidence they require to reduce that uncertainty.
If you only compare timelines, you miss the point. The pathway determines how much proof you must generate to justify patient safety.
Let’s break it down clearly.
The Real Comparison: Risk Tolerance
1. 510(k): Lowest Regulatory Uncertainty
The 510(k) pathway is built on one core concept: substantial equivalence.
FDA asks:
Is there an existing legally marketed device (predicate)?
Does your device have the same intended use?
Are technological differences minimal and not introducing new risks?
If the answer is yes, FDA does not require you to prove safety and effectiveness from scratch. You only need to demonstrate that your device is no riskier than what is already on the market.
Risk Tolerance Profile:
Low-to-moderate risk
Established regulatory history
Controlled technological differences
Evidence Expectation:
Bench testing
Performance validation
Possibly limited clinical data
FDA is tolerating some uncertainty—but only because precedent exists.
2. De Novo: Moderate Uncertainty, Managed Risk
De Novo applies when:
No predicate device exists
The device is novel
The risk level is still low-to-moderate
Here, FDA is willing to accept innovation—but only if risks can be clearly identified and mitigated.
Risk Tolerance Profile:
Moderate uncertainty
First-of-a-kind technologies
Risk controls must be clearly defined
Evidence Expectation:
More extensive bench testing
Often clinical evidence
Strong risk management documentation
FDA is not relying on precedent here. They are relying on your evidence.
3. PMA: Minimal Uncertainty Tolerance
Premarket Approval (PMA) is required when:
The device is high risk
It sustains or supports life
It is implanted long-term
Failure could cause serious harm
Here, FDA has very low tolerance for uncertainty.
Risk Tolerance Profile:
High patient impact
Significant harm potential
Limited room for ambiguity
Evidence Expectation:
Extensive preclinical testing
Well-controlled clinical trials
Comprehensive benefit-risk analysis
FDA requires strong, independent evidence of safety and effectiveness. Not similarity. Not plausibility. Proof.
What This Means Strategically
Choosing a pathway is choosing how much uncertainty your company must eliminate.
510(k): Reduce uncertainty by showing similarity
De Novo: Reduce uncertainty by demonstrating controlled innovation
PMA: Eliminate uncertainty through rigorous clinical proof
The higher the risk tolerance required, the more capital and time you must invest.
This is not about moving fast.
It is about aligning your device risk with the correct evidence burden.
The Most Common Founder Mistake
Many teams ask:
“Which pathway is fastest?”
The better question is:
“Which pathway reflects the true risk profile of this device?”
If the device risk is underestimated, the pathway will eventually shift.
And pathway shifts are expensive.
Where Kandih Comes In
This is where Kandih Group supports teams with structured comparative pathway analysis.
Kandih’s process includes:
Defining and pressure-testing intended use
Conducting classification and regulatory code assessments
Evaluating predicate strength and technological differences
Identifying hidden risk escalators
Comparing viable pathways side-by-side
Modeling cost, timeline, and capital exposure under each pathway
Instead of defaulting to the “fastest” route, teams make evidence-based pathway decisions aligned with FDA risk logic.
That protects:
Development timelines
Investor confidence
Capital allocation
Long-term strategy
Bottom Line
The difference between 510(k), De Novo, and PMA is not just procedural.
It is about how much uncertainty FDA is willing to accept—and how much proof you must provide.
Speed is a byproduct.
Risk tolerance is the driver.
Understanding that difference early prevents expensive pivots later.
References
FDA – Premarket Notification 510(k)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-notification-510k
FDA – De Novo Classification Process
https://www.fda.gov/medical-devices/premarket-submissions/de-novo-classification-request
FDA – Premarket Approval (PMA)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma
FDA – Classify Your Medical Device
https://www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device