The Complaint-to-CAPA Mapping Checklist
Do You Trend Complaints Before CAPA?
January 22, 2026
FDA Expects CAPAs to Feed Design Controls
January 26, 2026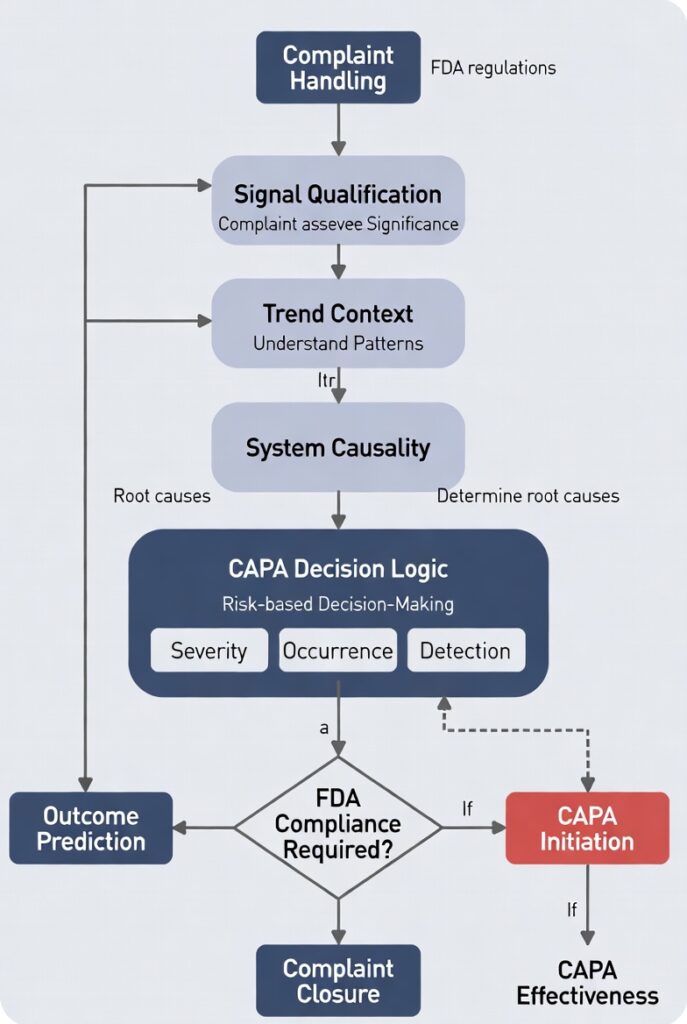
CAPA Survival Playbooks — Kandih Bioscience
Complaint-to-CAPA mapping is a risk-interpretation system, not an administrative checklist.
FDA evaluates whether complaint signals are consistently interpreted, escalated, or deferred based on documented risk logic. If the linkage between complaint and CAPA cannot be explained deterministically, FDA treats it as loss of governance—regardless of documentation completeness.
Why “End-of-Process” Checklists Fail Inspections
If your checklist lives at the end of the process, FDA already knows it’s decorative.
Many organizations assume that if complaints are investigated and CAPAs are closed, the linkage is self-evident.
That assumption fails inspections.
From the perspective of the U.S. Food and Drug Administration, linkage is not implied—it must be demonstrated. Inspectors are not asking whether complaints can lead to CAPA. They are testing whether your system predictably decides when they must, using risk logic rather than discretion.
The FDA Systems Perspective
Complaints generate signals
CAPA restores control
Between them must exist a deterministic mapping layer that answers—consistently and prospectively:
What signal did we observe?
What does it mean at the system level?
Why did it require—or not require—CAPA?
How was that decision validated over time?
If this logic is implicit, tribal, or reconstructed after the fact, FDA interprets it as loss of governance.
What FDA Inspectors Actually Evaluate
Inspectors are not asking for a prettier checklist.
They are stress-testing your decision architecture.
They look for evidence that:
Complaint signals are aggregated and trended before escalation
CAPA triggers are risk-based and reproducible, not reviewer-dependent
Similar complaints lead to similar outcomes, or differences are defensible
The absence of CAPA is justified using documented risk rationale
CAPA effectiveness is verified by post-implementation signal behavior, not closure dates
When these elements are missing, FDA sees system weakness—even when every record is complete.
The Complaint-to-CAPA Mapping Checklist (System Version)
This is not a to-do list.
It is a diagnostic map that must be answerable before CAPA decisions are made.
1. Signal Qualification
What failure mode does this complaint represent?
Is the signal known, emerging, or novel?
Has severity, use condition, or population shifted?
If this is unclear, CAPA timing will be indefensible.
2. Trend Context
Is this complaint part of a detectable pattern across time, product, or process?
Does the trend indicate stability or drift?
What threshold signals loss of control?
If trending happens after CAPA, the decision was not risk-informed.
3. System Causality Hypothesis
Which control failed—or never existed?
Is the issue localized or systemic?
Could the same failure occur elsewhere?
If causality stops at “use error,” FDA assumes analysis stopped prematurely.
4. CAPA Decision Logic
Why does this signal require CAPA—or why does it not?
What risk is being accepted if CAPA is not opened?
Would the same signal receive the same decision next month?
Inconsistent answers signal governance failure.
5. Outcome Prediction
What signal behavior should change if the decision is correct?
How will recurrence risk be measured?
What data confirms restored control?
Without prediction, effectiveness cannot be demonstrated.
The Regulatory Insight (Say This Plainly)
FDA does not cite missing checklists.
FDA cites systems that cannot explain their decisions.
A complaint-to-CAPA mapping failure is interpreted as:
Inadequate analysis of quality data
Subjective or inconsistent CAPA initiation
Ineffective corrective action
Weak management oversight
Administrative closure may look complete.
Regulatory effectiveness is judged by signal behavior over time.
The Design Principle (Your One Takeaway)
Design complaint-to-CAPA mapping as explicit decision logic, not procedural compliance.
If your system cannot clearly articulate:
What signal crossed which risk boundary, and
Why CAPA was the correct—or incorrect—control response,
then the mapping does not exist. It is being inferred after the fact.
FAQ
Q: Does FDA require a complaint-to-CAPA checklist?
A: FDA does not require a specific checklist. FDA requires consistent, risk-based decision logic that explains when complaints do or do not require CAPA.
Q: Can a complaint be closed without CAPA?
A: Yes—but only if the absence of CAPA is defensible using documented risk rationale and trend data.
Q: What does FDA look for when CAPA is not opened?
A: Evidence that complaint trends were analyzed and that control was still intact.
Q: Why do FDA observations cite “failure to analyze quality data”?
A: Because complaint-to-CAPA mapping logic was missing, inconsistent, or retrospective.
If your team cannot walk an inspector through complaint-to-CAPA decisions without narrative reconstruction, your system is operating on memory—not design.
That is precisely where diagnostic audits, CAPA remediation, and quality architecture review restore predictability.
If your CAPA system can’t explain itself to an inspector, it’s not designed yet.
References
21 CFR §820.100 — Corrective and Preventive Action (Medical Devices)
Establishes CAPA as a system for analyzing quality data, investigating causes, implementing actions, and verifying effectiveness—not as a paperwork exercise.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820/section-820.100
21 CFR §820.198 — Complaint Files (Medical Devices)
Defines complaints as quality data inputs that must be evaluated for failure modes, risk, and potential systemic issues—directly feeding CAPA expectations.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820/section-820.198
FDA Compliance Program Guidance Manual (CPGM) 7346.832 — QSIT
Describes how FDA investigators trace complaints, deviations, and trends into CAPA to assess system-level effectiveness and management control.
FDA Guidance: Quality Systems Approach to Pharmaceutical CGMP Regulations
Frames CAPA and investigations as part of an integrated quality system focused on signal detection, risk interpretation, and sustained control.
https://www.fda.gov/media/71023/download
21 CFR §211.192 — Production Record Review & Investigations (Drugs)
Requires thorough investigation of discrepancies and failures, forming the legal basis for CAPA-equivalent actions when systemic issues are identified.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211/section-211.192
21 CFR §211.198 — Complaint Files (Drugs)
Establishes complaint handling as a quality data source requiring evaluation for root cause and corrective action—not simple case closure.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211/section-211.198
ICH Q10 — Pharmaceutical Quality System
Positions CAPA within a closed-loop quality system emphasizing management responsibility, risk-based decision-making, and continual improvement.
https://database.ich.org/sites/default/files/Q10_Guideline.pdf
ICH Q9(R1) — Quality Risk Management
Defines how signals should be interpreted, escalated, and controlled using structured risk logic—directly applicable to complaint-to-CAPA mapping.
https://www.fda.gov/media/177720/download
FDA Warning Letters (Public Database)
Repeatedly cite failures to analyze complaint trends, initiate CAPA, and demonstrate effectiveness—reinforcing FDA’s enforcement logic around signal interpretation.

{kind=link}
{kind=link}
{kind=link}