How Early FDA Misalignment Increases Development Costs

Can Regulatory Strategy Begin Before a Prototype Exists?
February 11, 2026Why Investors Can Tell When Regulatory Was an Afterthought
February 13, 2026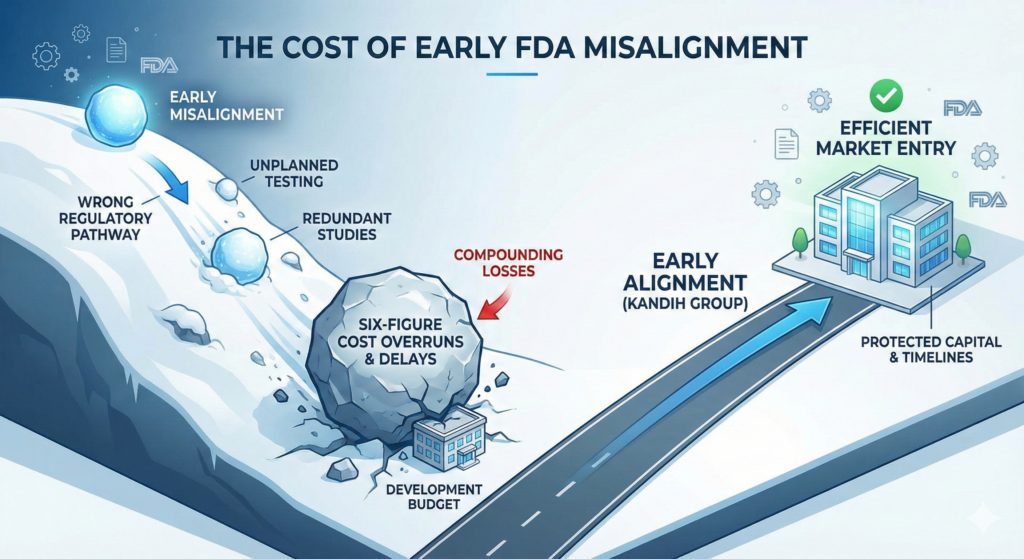
Simple truth: small regulatory misalignments early in development don’t stay small. They compound—quietly at first, then catastrophically—into six- and seven-figure problems later.
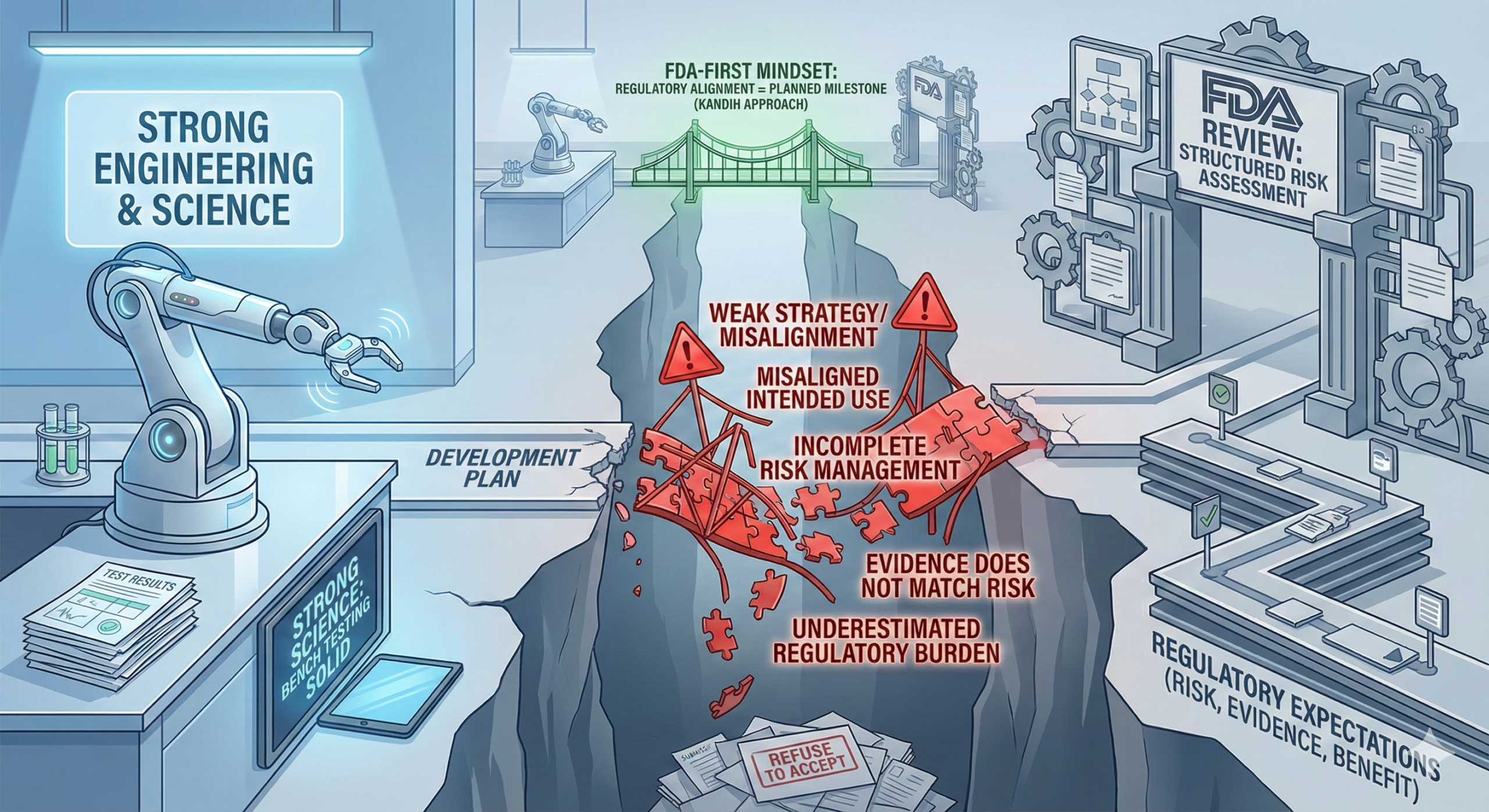
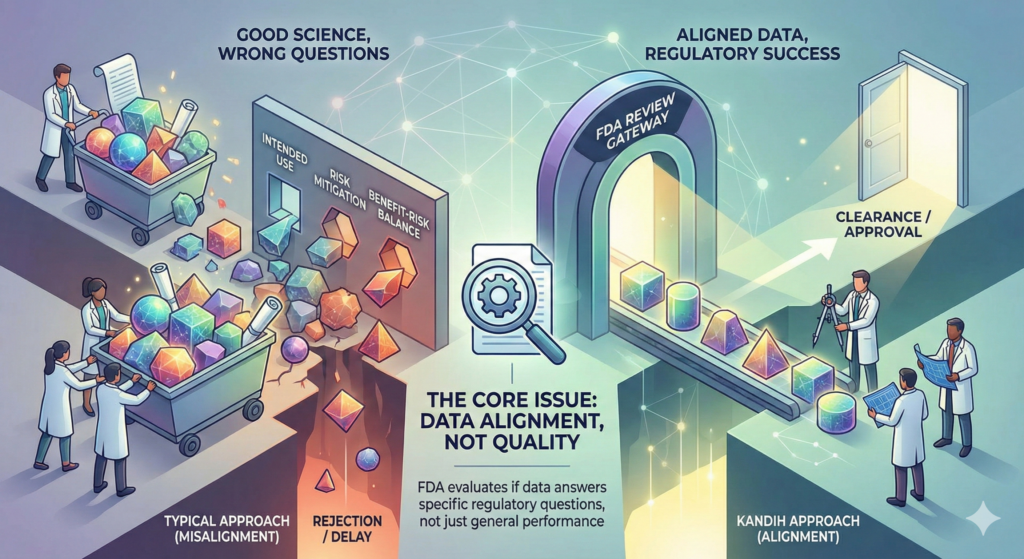
Most medical device cost overruns are not caused by bad engineering. They are caused by early FDA misalignment that went unnoticed or unchallenged.
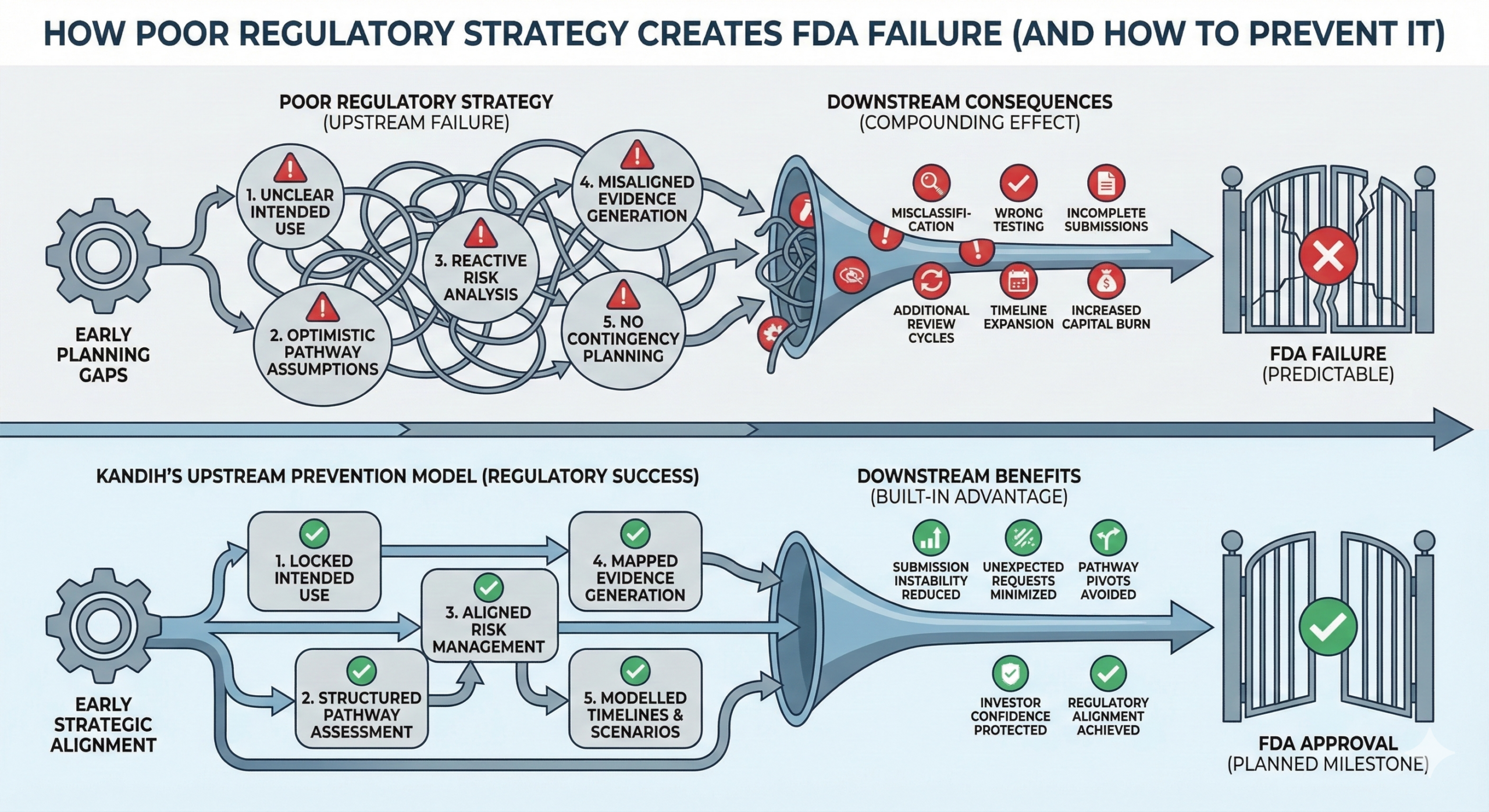
How Small Misalignments Snowball Into Big Losses
1. A Slightly Off Intended Use Becomes a New Regulatory Pathway
An intended use that is too broad, too clinical, or poorly worded can:
Push a device into a higher risk classification
Eliminate a viable 510(k) pathway
Force a De Novo or PMA route later
At the idea stage, this may feel like a wording issue. Downstream, it can mean a completely different development program.
Cost impact:
Additional testing: $500,000–$2M
Timeline impact: 12–24 months
2. Early Design Decisions Trigger Unplanned Testing
Choosing a material, software feature, or energy source without FDA context often leads to:
Unexpected biocompatibility testing
Higher software validation burden
Additional electrical or electromagnetic compatibility testing
Expanded human factors studies
None of these are “mistakes” on their own—but together they add up fast.
Cost impact:
Incremental testing: $250,000–$1M
Engineering rework and delays
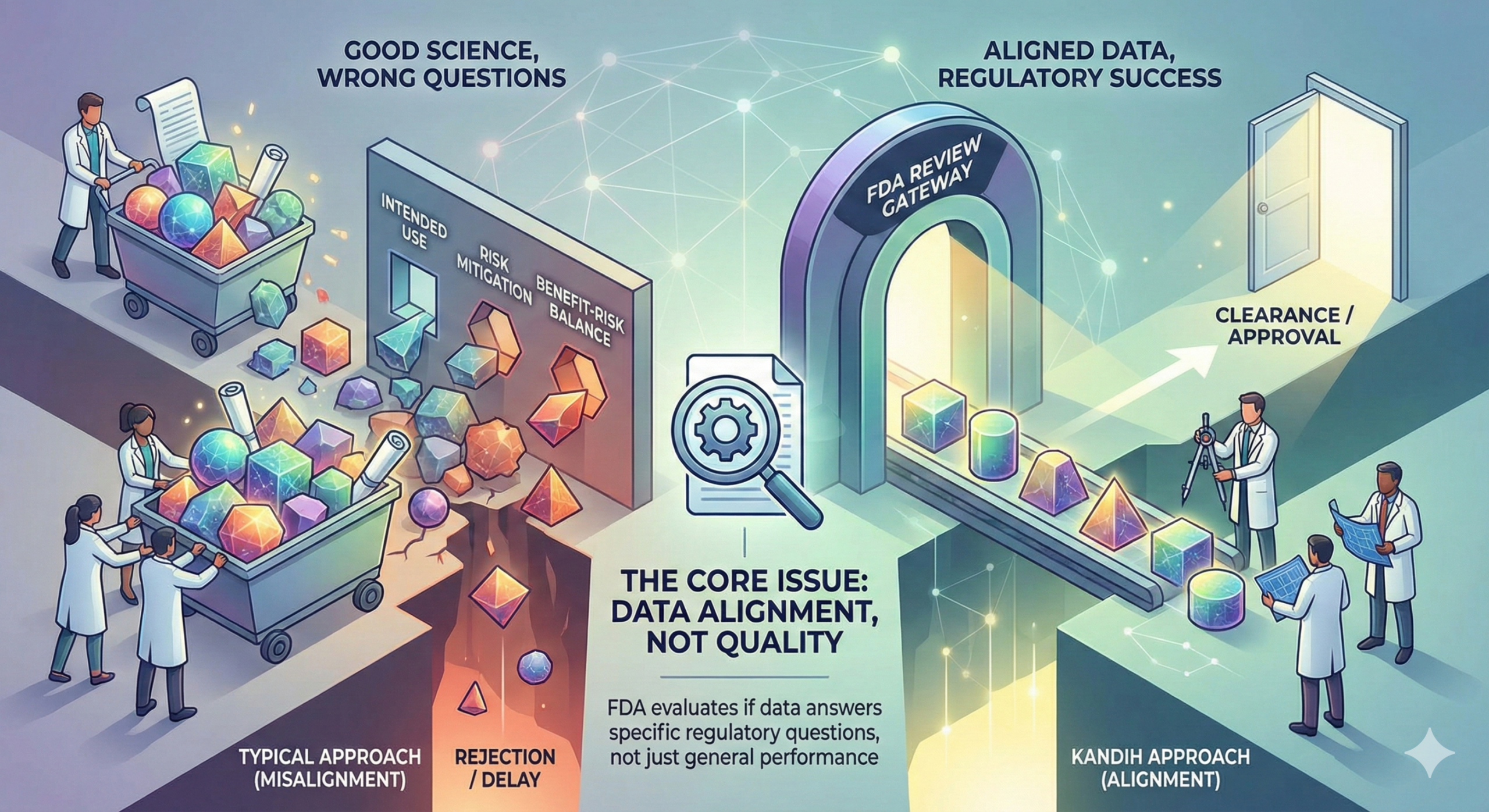
3. Misaligned Evidence That FDA Won’t Accept
When regulatory strategy is late, teams often generate data that:
Is not tied to identified risks
Uses the wrong endpoints
Cannot be traced back to design inputs
This leads to the worst outcome of all: valid-looking data that FDA cannot use.
Cost impact:
Repeating studies: $300,000–$1.5M
Lost time explaining, defending, and redoing work
FDA evaluates evidence through a structured benefit-risk lens. If your data does not align with that logic, it simply does not count.
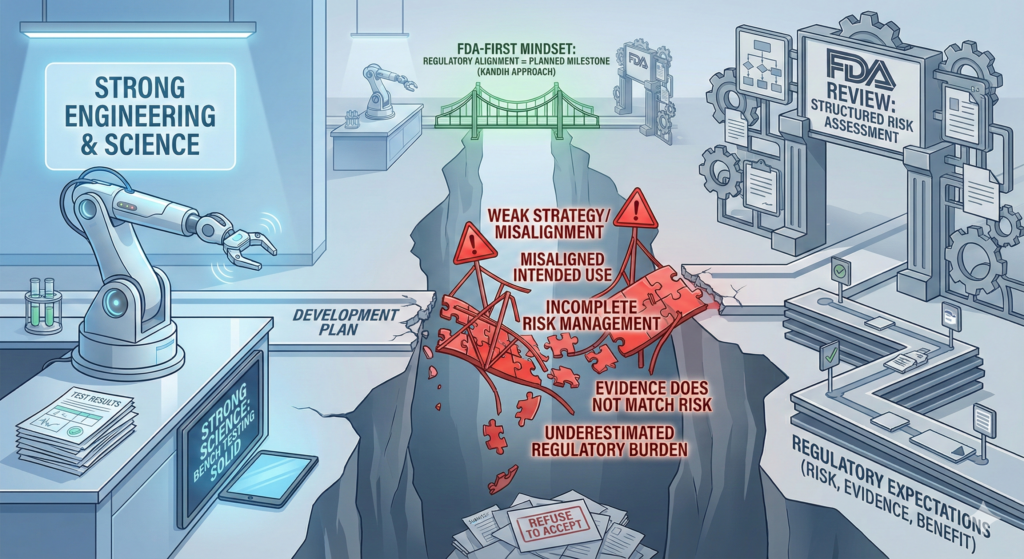
4. Documentation Gaps Multiply Review Cycles
Early misalignment often shows up later as:
Weak design history files
Missing rationale for testing decisions
Inconsistent risk documentation
This triggers repeated information requests during FDA review and increases the chance of refusal-to-accept decisions.
Cost impact:
Prolonged FDA review
Burned internal and external resources
Missed market windows
This is where regulatory debt comes due—with interest.
The Compounding Effect (Reality Check)
Across medical device programs, early FDA misalignment commonly leads to:
30–60% development cost overruns
1–3 years of avoidable delay
Reduced investor confidence
Loss of first-mover advantage
None of this is visible at the whiteboard stage—but all of it is baked in there.
Where Kandih Comes In
This is exactly what Kandih Group is designed to stop early.
Kandih identifies misalignment before it becomes expensive by:
Pressure-testing intended use and claims against U.S. Food and Drug Administration expectations
Mapping early design choices to regulatory consequences
Aligning risk classification, evidence planning, and development strategy
Flagging regulatory “cost multipliers” before capital is deployed
Helping teams build once—correctly
Instead of paying to fix problems later, founders invest a fraction upfront to avoid them entirely.
Bottom Line
Early FDA misalignment is not a paperwork issue.
It is a cost multiplier.
The cheapest regulatory decision is the one you make early—before design choices harden and evidence is generated.
That’s how Kandih helps innovators protect capital, timelines, and credibility.
References
FDA – Classify Your Medical Device
https://www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device
FDA – Design Controls for Medical Devices
https://www.fda.gov/files/drugs/published/Design-Controls—Devices.pdf
FDA – Refuse to Accept Policy for 510(k)s
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/refuse-accept-policy-510ks
FDA – Benefit-Risk Factors for Medical Devices
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/factors-consider-regarding-benefit-risk-medical-device-product-availability-compliance-and

{kind=link}
{kind=link}
{kind=link}