Design Controls Pain Points: R&D vs. Regulatory Disconnects
BIOCOMPATIBILITY • REGULATORY STRATEGY • INVESTOR INSIGHT
June 8, 2026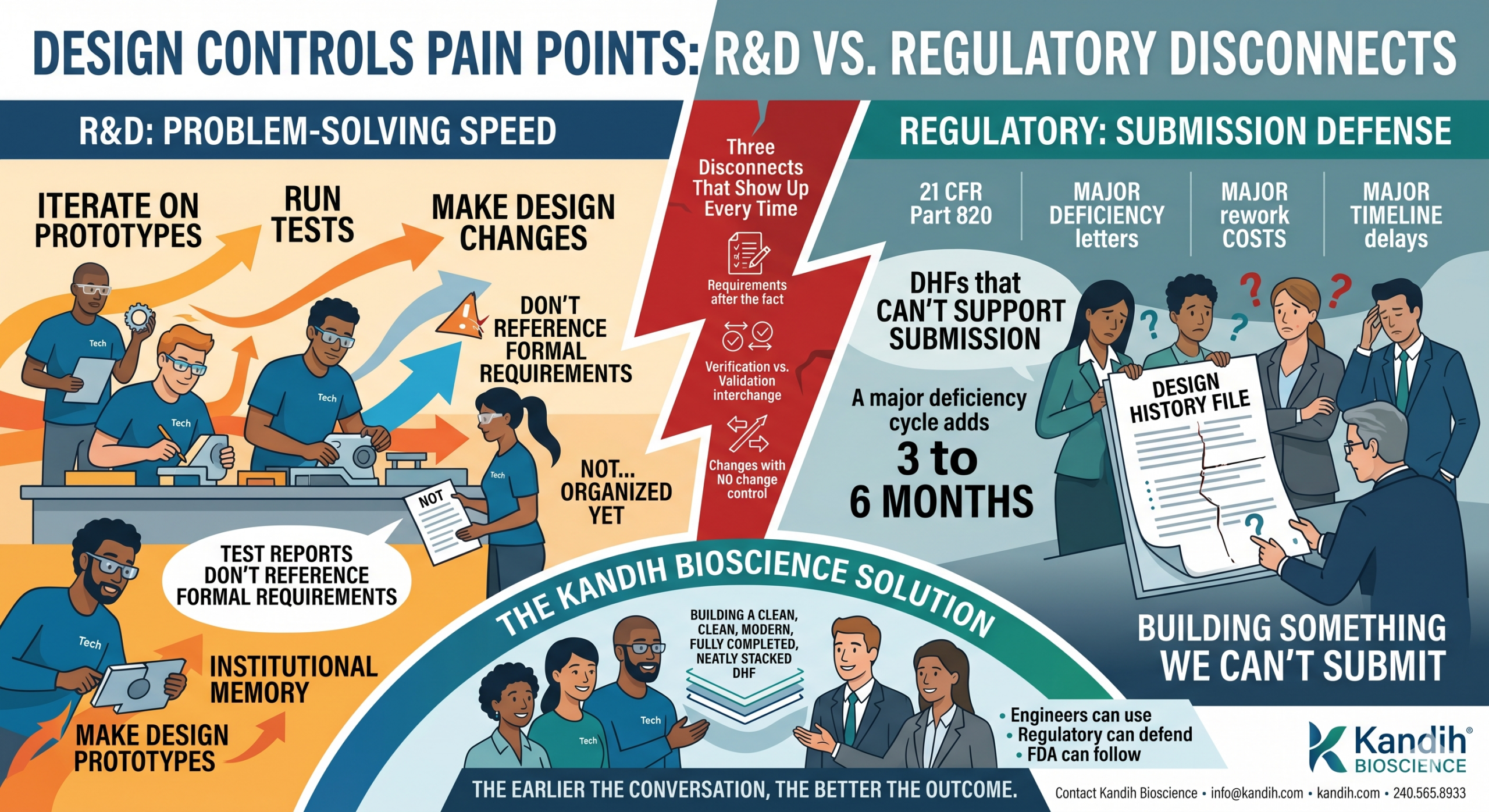
By Kandih Bioscience • Regulatory Strategy Series
From r/medicaldevices: “Our R&D team thinks design controls are bureaucratic box-ticking. Our regulatory lead says we’re building something we can’t submit. They’re in the same room and they’re not speaking the same language. Has anyone actually fixed this?”
That post got 200 comments in 48 hours.
Because every medical device founder, regulatory lead, and engineer who has been through a 510(k) or PMA submission recognized exactly what that person was describing.
The tension between R&D and regulatory is one of the most talked-about, least-solved problems in medical device development. And it almost always comes back to the same place: design controls.
Design controls are the system FDA requires you to use to plan, build, and document your device. Done right, they’re a roadmap. Done wrong — or ignored until submission — they become the most expensive rework you’ll ever do.
This is the story of how that disconnect happens, what it costs, and what it actually looks like to fix it.
What Design Controls Actually Are (And Why Engineers Resist Them)
Let’s start with what design controls are, because the name doesn’t help anyone.
Under 21 CFR Part 820 — FDA’s Quality System Regulation — design controls are a set of documented activities you’re required to follow when developing a device. They cover the full arc of development: from the moment you define what your device needs to do, through design and testing, all the way to release.
The system has a specific vocabulary:
- Design inputs: what the device must do — the requirements
- Design outputs: what you actually built — the specs, drawings, and software that define the device
- Design verification: did we build it right? — testing that the outputs meet the inputs
- Design validation: did we build the right thing? — testing that the device works for the intended user in the intended environment
- Design history file (DHF): the complete, organized record of all of the above
On paper, this looks reasonable. In practice, it’s where the friction starts.
Most engineers didn’t go to school to write documentation. They went to school to solve problems. When a regulatory person walks in and says ‘you need to trace every design requirement to a test result,’ the instinct is to hear: ‘stop building and start filling out forms.’
That instinct isn’t wrong. It’s just pointed at the wrong target.
The documentation doesn’t slow down the building. The lack of a shared plan does.
For founders: if your engineering team and your regulatory lead are having this fight, it’s usually not a personality conflict. It’s a process gap. They don’t have a shared framework for how design decisions get made and documented. That gap is fixable — but it compounds the longer you wait.
The Moment the Disconnect Becomes a Crisis
Here’s how the story usually goes.
A device company starts development. The engineering team moves fast — iterating on prototypes, running tests, making design changes. Progress is happening. Everyone is excited.
Somewhere around month six or eight, someone — usually the regulatory lead, sometimes an outside consultant, sometimes a potential investor asking due diligence questions — asks to see the design history file.
And the answer is some version of: ‘We have everything. It’s just not… organized yet.’
What they actually have is a folder full of test reports that don’t reference any formal requirements. Prototype iterations that were never documented as design changes. Bench test data that proves something was measured but not what requirement it was measuring against. Meeting notes. Email threads. Institutional memory that lives entirely in people’s heads.
None of that is a design history file. All of it has to become one before you can submit.
For investors: when you’re evaluating a device company and they tell you they’re ‘90 days from submission’ — ask to see their DHF. If the answer is hesitation, backpedaling, or a promise that it’s ‘almost done’, that 90-day estimate almost certainly doubles. The cost of reconstructing a design history from scattered records is measured in months, not weeks.
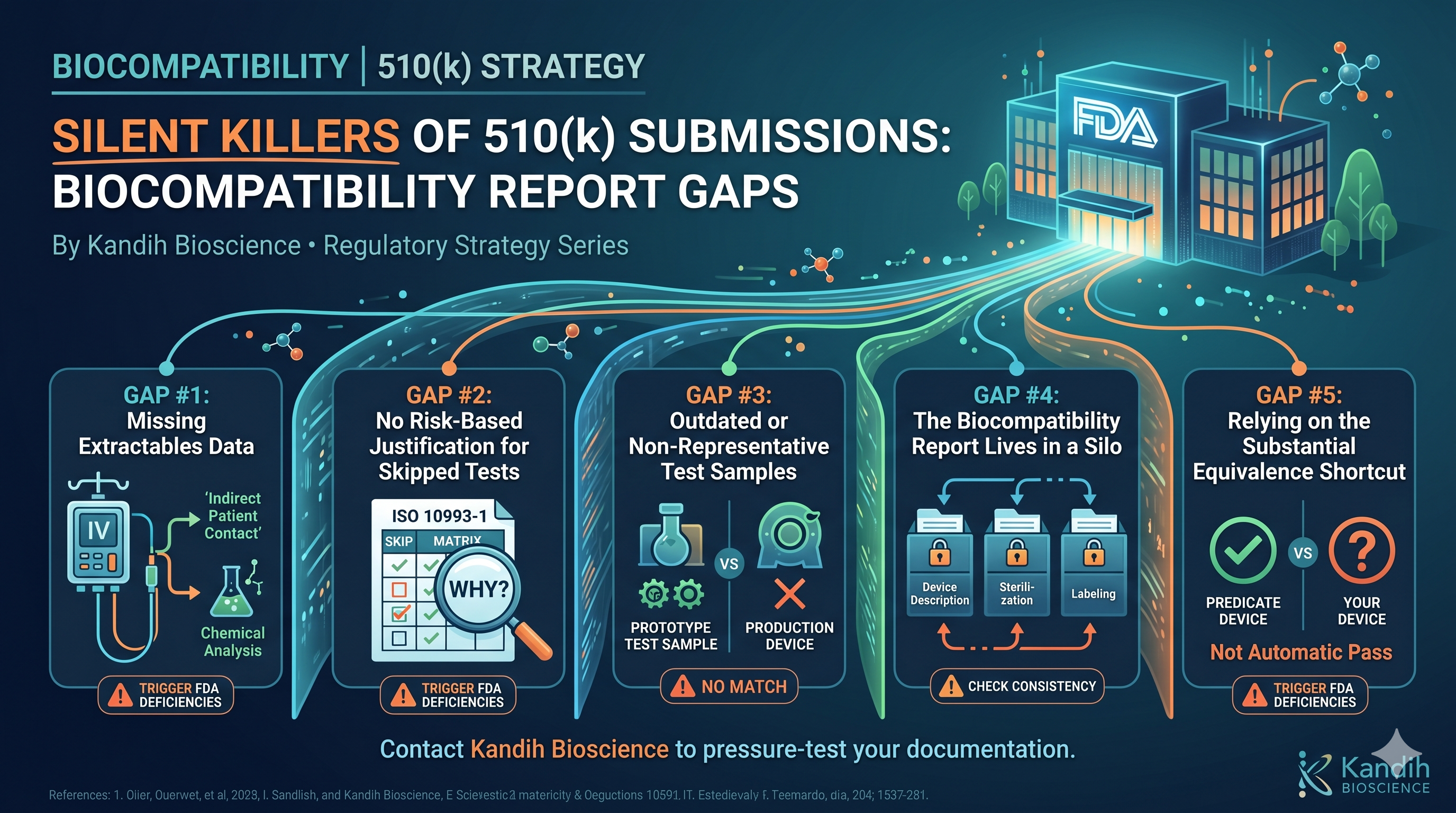
The Three Disconnects That Show Up Every Time
After years of working with device companies on design controls, the same three breakdowns appear in almost every case. They’re different problems, but they all feed the same outcome: a DHF that can’t support submission.
Disconnect #1: Requirements written after the fact.
Design inputs are supposed to come first. They define what the device must accomplish — performance thresholds, safety requirements, user needs, environmental conditions. Everything that gets built should be traceable back to a requirement.
What usually happens: requirements get written after the design is mostly complete, reverse-engineered from what was already built. Engineers call this ‘writing down what we already know.’ Regulators call it untraceable.
FDA’s concern isn’t that the device doesn’t work. It’s that you can’t demonstrate a controlled process. A device that performs well but has no documented design process doesn’t give FDA confidence that the process was controlled — and that’s what the regulation requires.
Disconnect #2: Verification and validation used interchangeably.
These are two different activities. Verification tests whether you built the device to spec. Validation tests whether the device actually works for the user in real conditions.
A bench test that confirms a component meets a dimensional tolerance is verification. A usability study that confirms a nurse can operate the device without making an error in a realistic clinical setting is validation.
Companies that run bench tests and label them ‘V&V’ without distinguishing which is which end up with a submission gap. FDA will ask specifically for evidence of clinical or simulated-use validation. Bench data alone doesn’t answer that question.
Disconnect #3: Design changes with no change control.
Devices change during development. Materials get swapped. Dimensions shift. Software gets updated. Every one of those changes is a design change, and every design change is supposed to be evaluated for its impact on the device’s safety and performance.
What actually happens: changes happen organically, logged (if at all) in engineering notebooks or version control systems that aren’t connected to the formal design record. By submission time, the device that’s been tested might not match the device that’s been described — because nobody tracked the delta.
FDA’s auditors are specifically trained to find these gaps. A design change that isn’t documented is a Form 483 observation waiting to happen.
What This Costs You as a Founder
Let’s be direct about the financial picture.
The most common outcome of a design controls gap isn’t a rejected submission. It’s a delayed one. FDA sends a major deficiency letter. You go back and reconstruct the documentation. You resubmit. FDA restarts its review clock.
A single major deficiency cycle typically adds 3 to 6 months to a 510(k) timeline. For a PMA, the impact is larger.
Three to six months of runway. Three to six months of salaries, facility costs, and burn rate. Three to six months of delayed revenue. Three to six months before you can start the commercial conversations your investors are waiting for.
The design controls work itself — if you do it at the beginning of development — takes weeks. Reconstructing it at the end, under submission pressure, takes months. The work is the same. The timeline is completely different.
The founders who treat design controls as a development tool — not a submission requirement — consistently hit shorter clearance timelines. Not because they’re more compliant. Because they don’t have to do the same work twice.
What Actually Fixes the R&D vs. Regulatory Disconnect
The fix is not more documentation. It’s an earlier conversation.
The R&D team and the regulatory function need to agree — before the first prototype is built — on three things:
- What are the design inputs? What must this device do, for whom, under what conditions, with what safety requirements? This isn’t a regulatory question. It’s an engineering question with regulatory implications.
- How will changes be tracked? Not in retrospect. As they happen. The system doesn’t need to be complicated — it just needs to exist and be used.
- What does ‘tested’ mean? Every test report should reference the requirement it’s testing against. If your engineers don’t know what requirement a test addresses, the test result can’t go in a DHF that FDA can follow.
None of this requires a separate regulatory process running parallel to engineering. It requires a shared vocabulary and a simple, consistent habit: connect every decision to a requirement, and document the connection.
That habit, started early, is what separates a 6-month submission process from an 18-month one.
Where Kandih Bioscience Comes In
The R&D vs. regulatory tension is real. But it’s not inevitable.
At Kandih Bioscience, we work with device companies at the point where this friction is highest — often somewhere between initial prototyping and the first serious submission conversation. We help teams build a design controls framework that engineers can actually use, that regulatory can actually defend, and that FDA can actually follow.
That means:
- Structuring design inputs in a way that maps cleanly to your verification and validation activities
- Reviewing your existing records — test reports, change logs, meeting notes — and identifying what can be used, what needs to be supplemented, and what gaps remain
- Building a traceability matrix that connects requirements to outputs to test results, end to end
- Helping your team establish a change control habit that doesn’t slow engineering down but keeps the DHF current
If you’re a founder who has felt the friction between your engineering team and your regulatory function, we’ve seen it before. The gap is fixable. The question is whether you fix it now or six months from now, under submission pressure.
If you’re an investor looking at a device company and you want to understand the regulatory readiness of their design documentation, we can give you a plain-language assessment of where they stand.
Either way: the earlier the conversation, the better the outcome.
Contact Kandih Bioscience • info@kandih.com • kandih.com • 240.565.8933
References
1. FDA — Design Controls Guidance for Medical Device Manufacturers (21 CFR Part 820.30) (1997, updated 2023)
2. FDA — Quality System Regulation: 21 CFR Part 820 (current)
4. FDA — Design History File and Device Master Record Checklist (2014)
5. ISO 13485: Medical Devices — Quality Management Systems (2016)
6. FDA — Guidance on Verification and Validation for Software-Based Medical Devices (2005)
7. FDA — Human Factors and Usability Engineering — Guidance for Medical Device Manufacturers (2016)