Complaint → Weak Root Cause → Repeat 483

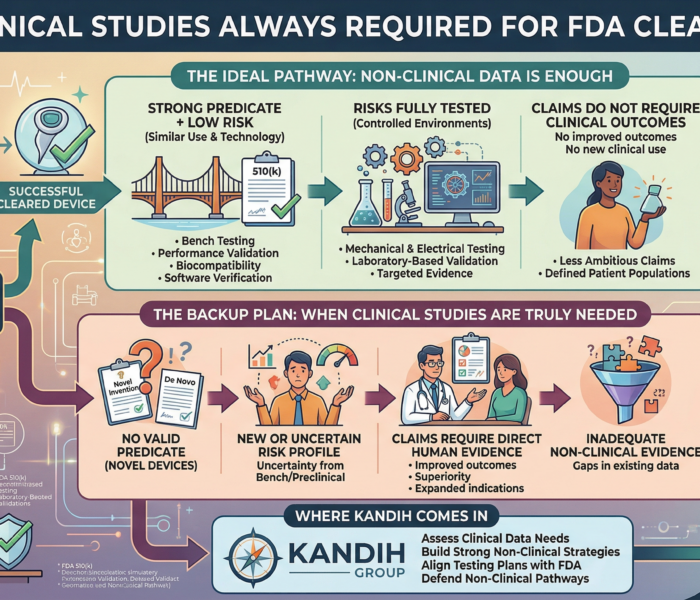
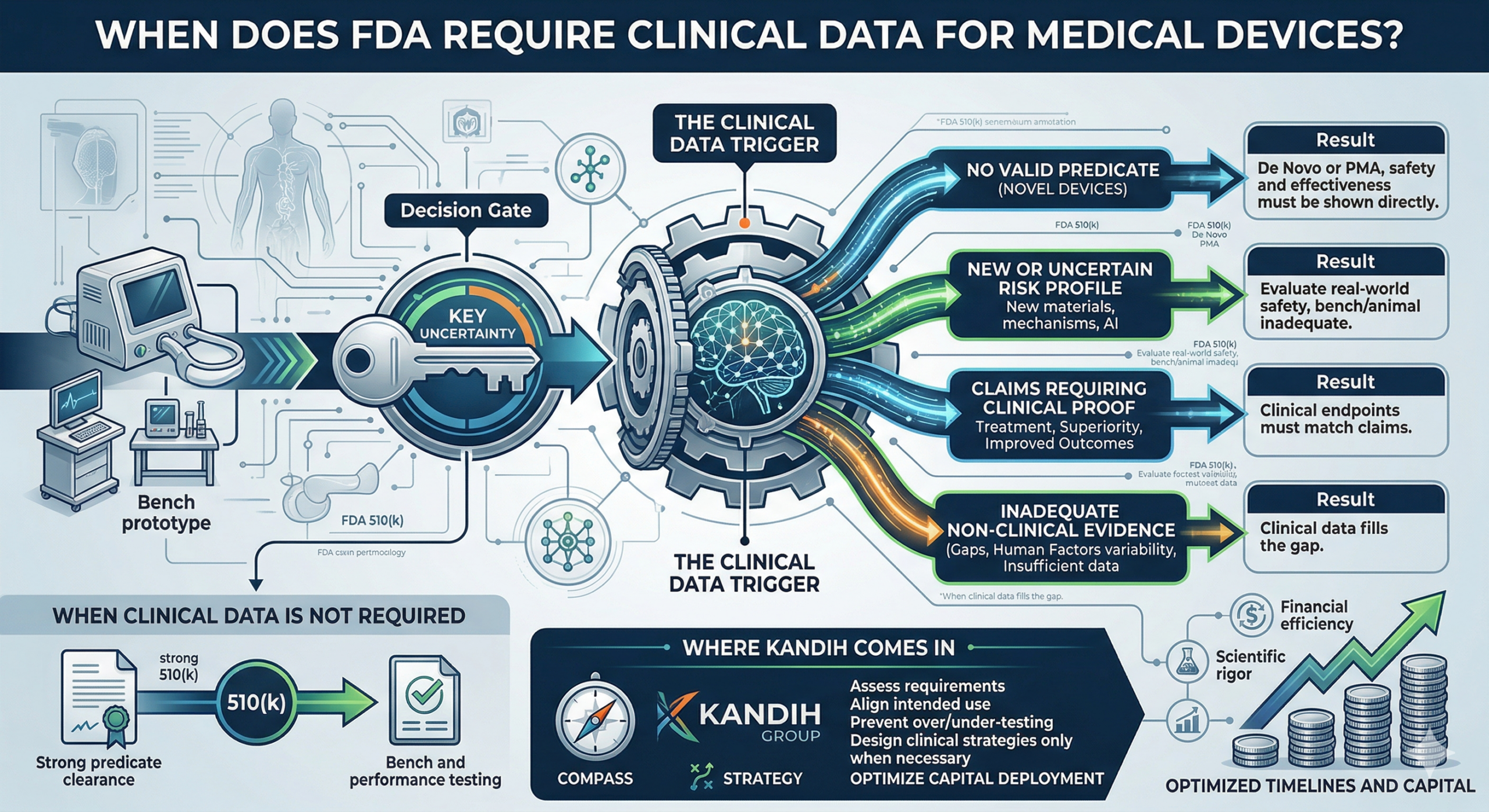
When Does a Complaint Legally Require CAPA?
January 20, 2026
Do You Trend Complaints Before CAPA?
January 22, 2026
CAPA Survival Playbooks — Kandih Bioscience
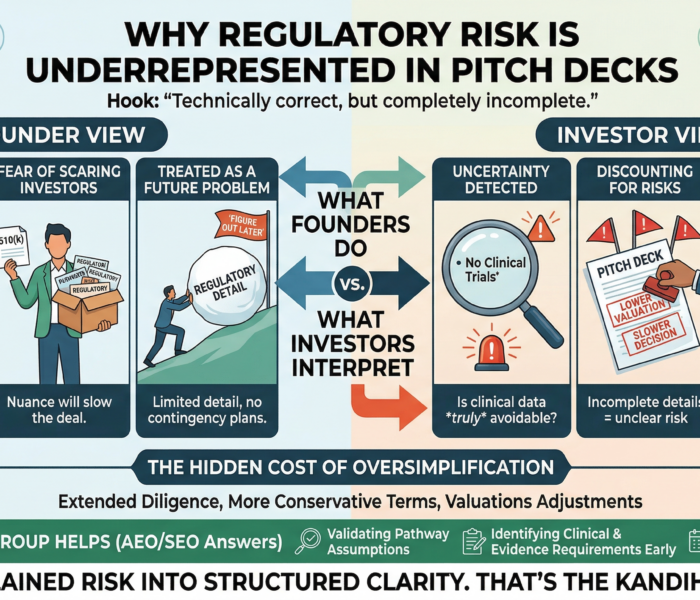
Repeat FDA 483 observations almost always mean the original root cause was wrong.
From an FDA enforcement perspective, recurrence equals misdiagnosis. If the root cause did not explain how the system failed—or why it could fail again—CAPA did not restore control, regardless of how clean the documentation looked.
Why “Tidy” Root Causes Trigger FDA Suspicion
If your root cause feels neat, contained, and emotionally satisfying, FDA already suspects it’s incomplete.
Repeat 483s rarely come from new failures.
They come from old signals that were misdiagnosed.
The assumption that keeps companies cited
Common belief:
“We investigated the complaint, identified the root cause, and closed the CAPA.”
FDA reality:
Closure is administrative. Control is regulatory.
The U.S. Food and Drug Administration does not evaluate whether you named a root cause.
FDA evaluates whether your system understood the failure well enough to prevent recurrence.
When the same observation returns, FDA is not seeing a documentation lapse.
FDA is seeing a diagnostic failure.
The FDA Systems View (Where Things Actually Break)
From an inspector’s perspective, the logic is simple and unforgiving:
Complaint → Root Cause → Control → Outcome
A repeat 483 tells FDA one thing with clarity:
The root cause did not explain the system failure.
Not partially.
Not informally.
Architecturally.
Weak root cause is not a CAPA execution issue.
It is a Causality Layer failure in the quality system.
What FDA Inspectors Are Actually Evaluating
Inspectors are not impressed by the tools used (5 Whys, fishbone, etc.).
They evaluate whether your root cause analysis:
Explains why the system allowed the failure
Identifies which control failed or never existed
Accounts for recurrence risk across similar processes
Leads to controls that change system behavior, not staff behavior
Predicts an outcome that can be verified over time
If the same issue reappears, FDA assumes the earlier root cause was symptomatic, not causal.
How Weak Root Cause Predictably Produces Repeat 483s
1. Root Cause Stops at the Human
“Operator error.”
“Failure to follow procedure.”
“Inadequate training.”
FDA interpretation:
Humans operate inside systems. If behavior is the cause, the system enabled it.
Stopping here tells FDA you don’t understand your own controls.
2. System Interfaces Are Ignored
Complaints implicate design limits, labeling, detection thresholds, handoffs, or incentives—but analysis stays local.
FDA interpretation:
You fixed the node, not the network. Expect recurrence elsewhere.
3. Controls Don’t Map Back to the Cause
Training completed. SOPs revised. No change to design, detection, validation, or workflow.
FDA interpretation:
The control layer is unchanged. CAPA is cosmetic.
4. Effectiveness Is Declared, Not Demonstrated
CAPA closed after a qualitative review. No trend analysis. No outcome shift.
FDA interpretation:
Administrative closure without proof of durable control equals ineffective CAPA.
That’s why the observation comes back—usually sharper.
The Regulatory Insight (Uncomfortable, but Accurate)
A repeat 483 is FDA stating:
Your previous root cause did not explain reality.
Not that it needed more detail.
Not that documentation was weak.
But that it failed to model the system well enough to control risk.
From FDA’s standpoint, recurrence equals misdiagnosis.
The Design Principle (Your One Takeaway)
Root cause must explain recurrence—not just the last event.
A defensible root cause can answer:
Why would this happen again elsewhere?
Which system control failed—or never existed?
How does the corrective action measurably reduce recurrence risk?
If it cannot, the analysis is narrative, not diagnostic.
And narrative root causes produce repeat 483s.
FAQ
Q: Why does FDA issue repeat 483s for the same issue?
A: Because the original root cause did not explain the system failure well enough to prevent recurrence.
Q: Is “operator error” an acceptable root cause to FDA?
A: Rarely by itself. FDA expects identification of the system control that allowed the error.
Q: Does closing a CAPA mean FDA considers it effective?
A: No. FDA evaluates effectiveness based on recurrence and outcome trends, not closure status.
Q: What does FDA assume when an issue reappears?
A: That the prior root cause was symptomatic, not causal.
If your organization has ever said, “We already addressed this last time,” that is a signal—not a defense.
That is the moment to reassess whether your complaint handling and CAPA system is designed to understand failure, not just document it.
If your CAPA system can’t explain itself to an inspector, it’s not designed yet.
References
21 CFR §820.100 — Corrective and Preventive Action (Medical Devices)
Establishes FDA’s expectation that CAPA must identify causes of nonconformities, implement systemic corrections, and verify effectiveness—not merely document actions.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820/section-820.100
21 CFR §820.198 — Complaint Files (Medical Devices)
Defines complaint handling as a critical input into CAPA and quality system control, not a standalone administrative function.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820/section-820.198
21 CFR §211.192 — Production Record Review (Drugs)
Requires investigation of unexplained discrepancies and failures, forming the legal backbone for CAPA expectations in pharmaceutical CGMP inspections.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211/section-211.192
21 CFR §211.198 — Complaint Files (Drugs)
Establishes complaint investigation as a trigger for systemic corrective action when quality or safety issues are identified.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211/section-211.198
ISO 13485:2016 — Medical Devices QMS
Requires documented root cause analysis and verification of CAPA effectiveness across the system lifecycle.
https://www.iso.org/standard/59752.html
FDA Warning Letters Database
Public enforcement record demonstrating how weak root cause and ineffective CAPA lead to repeat 483s and escalated enforcement.
FDA Form 483 Database
Illustrates how inspectors document repeated observations when prior CAPAs fail to prevent recurrence.

{kind=link}
{kind=link}
{kind=link}