Why “We’ll Try 510(k)” Is a Red Flag
What Is the Difference Between 510(k), De Novo, and PMA—Really?
February 23, 2026
The 100-Year Mirage: Why Modern Longevity is a Childhood Survival Story
February 25, 2026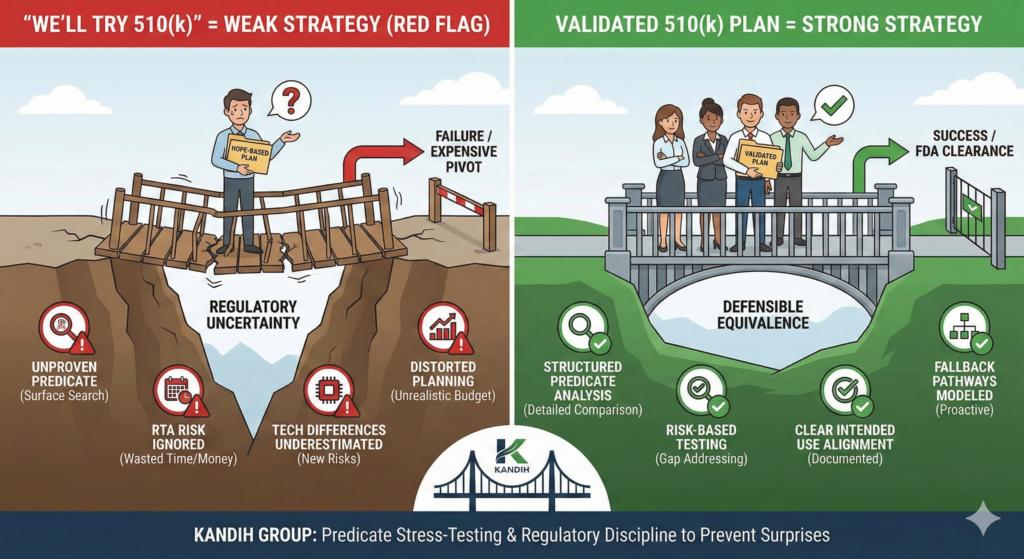
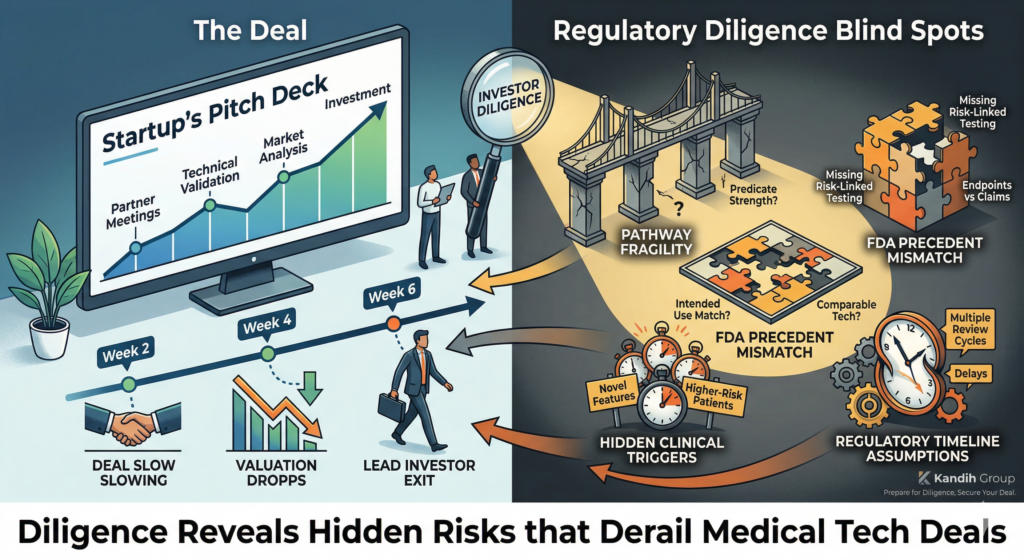
Short answer: when a team says, “We’ll try 510(k),” it usually means the regulatory strategy is not fully developed.
A 510(k) is not something you “try.” It is something you qualify for.
Under the framework of the U.S. Food and Drug Administration, the 510(k) pathway depends on a very specific standard: substantial equivalence to a legally marketed predicate device.
If that logic is not solid, the pathway collapses.
Why “We’ll Try 510(k)” Signals Weak Strategy
1. It Suggests Predicate Assumptions Haven’t Been Proven
A valid 510(k) requires:
Same intended use as the predicate
Similar technological characteristics
No new questions of safety or effectiveness
If those elements are not clearly documented and defensible, the application is vulnerable.
Saying “we’ll try” often means:
Predicate searches were surface-level
Technological differences were not deeply analyzed
Risk escalation wasn’t fully assessed
That is not a strategy. That is hope.
2. It Ignores the Risk of Refuse-to-Accept (RTA)
FDA can refuse to accept a 510(k) submission if:
Required elements are missing
Substantial equivalence is poorly justified
Testing does not align with technological differences
An RTA decision is not a minor setback. It costs:
Months of delay
Additional consulting fees
Investor confidence
“We’ll try 510(k)” does not account for this risk.
3. It Underestimates Technological Differences
Many teams believe that small changes do not matter.
But FDA evaluates whether differences:
Introduce new mechanisms of action
Change performance characteristics
Alter risk profile
If the device includes:
AI-based functionality
New materials
Expanded clinical claims
Different energy delivery
Those differences may disqualify 510(k).
This is where optimism becomes expensive.
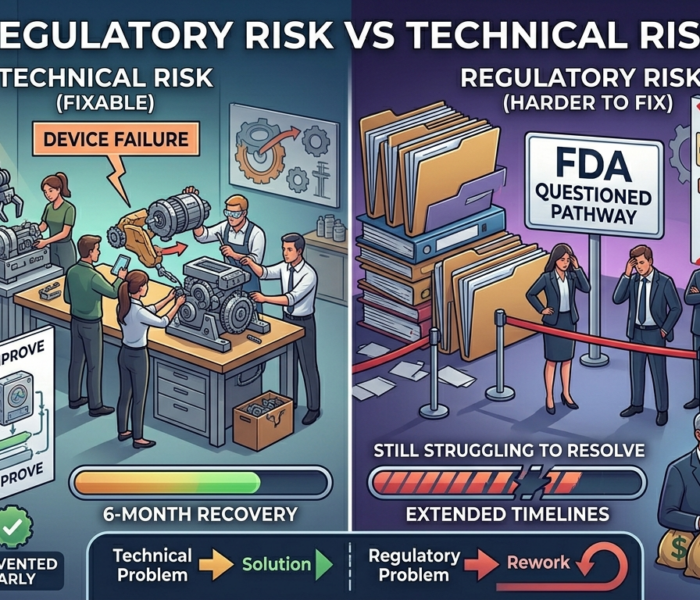
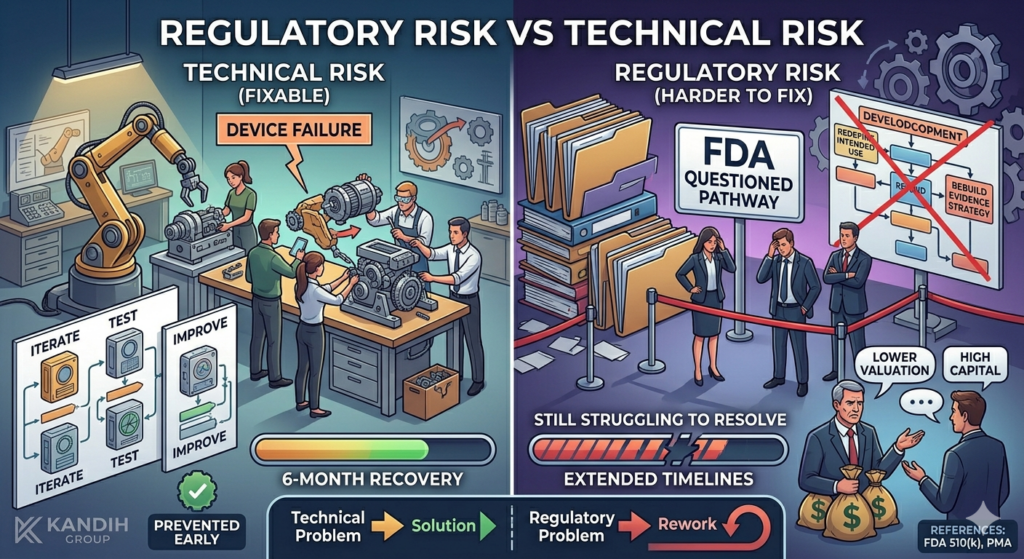
4. It Distorts Timeline and Budget Planning
If 510(k) is assumed but not validated, downstream planning becomes flawed:
Testing budgets are underestimated
Clinical study planning may be deferred
Investor projections become unrealistic
If the pathway later shifts to De Novo or PMA, cost exposure increases dramatically.
That shift rarely happens quietly. It happens during diligence or FDA review.
What a Strong 510(k) Strategy Looks Like
A defensible 510(k) plan includes:
Structured predicate identification
Detailed technological comparison
Risk analysis tied to differences
Testing plans directly addressing risk gaps
Clear intended use alignment
It does not rely on “similar enough.”
It relies on documented equivalence.
Where Kandih Comes In
This is where Kandih Group provides structured predicate stress-testing.
Kandih supports teams by:
Conducting deep predicate landscape analysis
Comparing technological characteristics line-by-line
Identifying hidden safety questions
Evaluating whether differences introduce new risk categories
Modeling fallback pathways if 510(k) fails
Building defensible substantial equivalence arguments
Instead of “trying” 510(k), teams know whether it is viable before capital is deployed.
That protects:
Development timelines
Investor confidence
Regulatory credibility
Bottom Line
“We’ll try 510(k)” signals uncertainty.
Strong regulatory strategy does not guess.
It validates.
If 510(k) is viable, it should be defendable.
If it is not, the pivot should happen early—not after submission.
That is how regulatory discipline prevents expensive surprises.
References
FDA – Premarket Notification 510(k)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-notification-510k
FDA – Refuse to Accept Policy for 510(k)s
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/refuse-accept-policy-510ks
FDA – Substantial Equivalence in Premarket Notifications (510(k))
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/substantial-equivalence-premarket-notifications-510k
FDA – Classify Your Medical Device
https://www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device


{kind=link}
{kind=link}
{kind=link}