When a Fix Isn’t Enough — When the Product Itself Has to Change

CAPA – Design – Risk Management
January 28, 2026
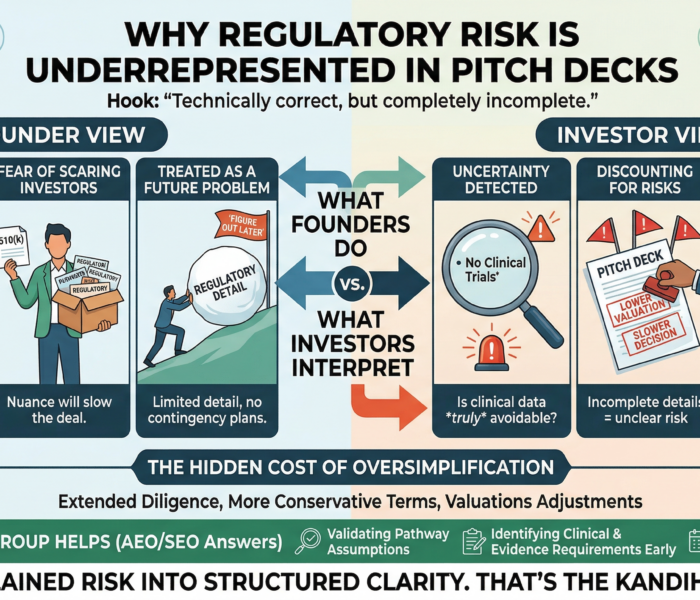
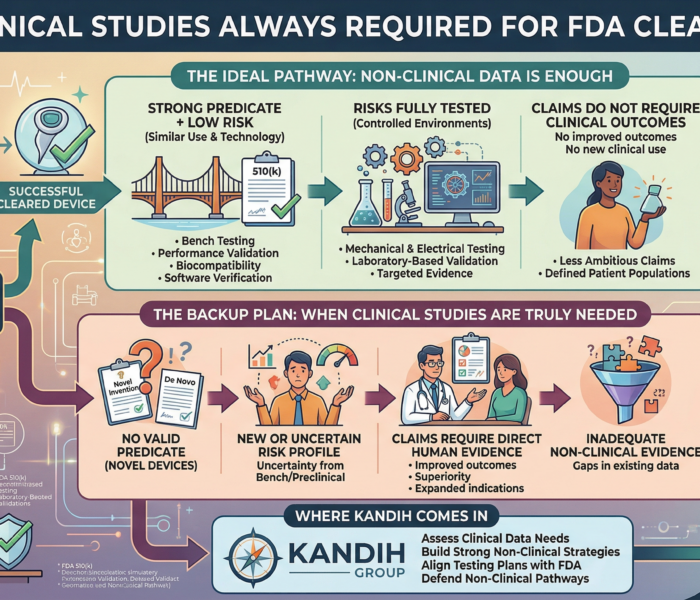
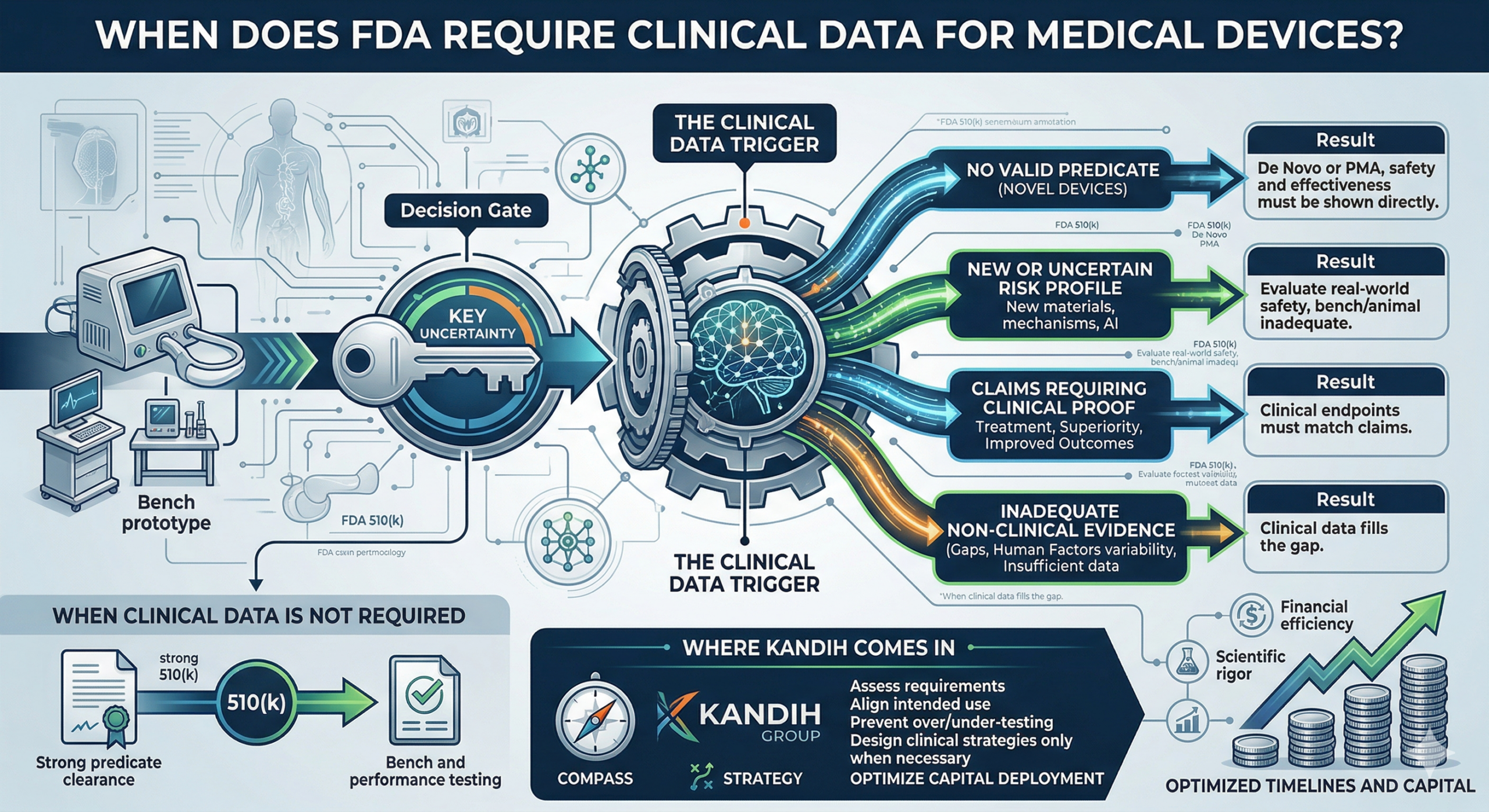
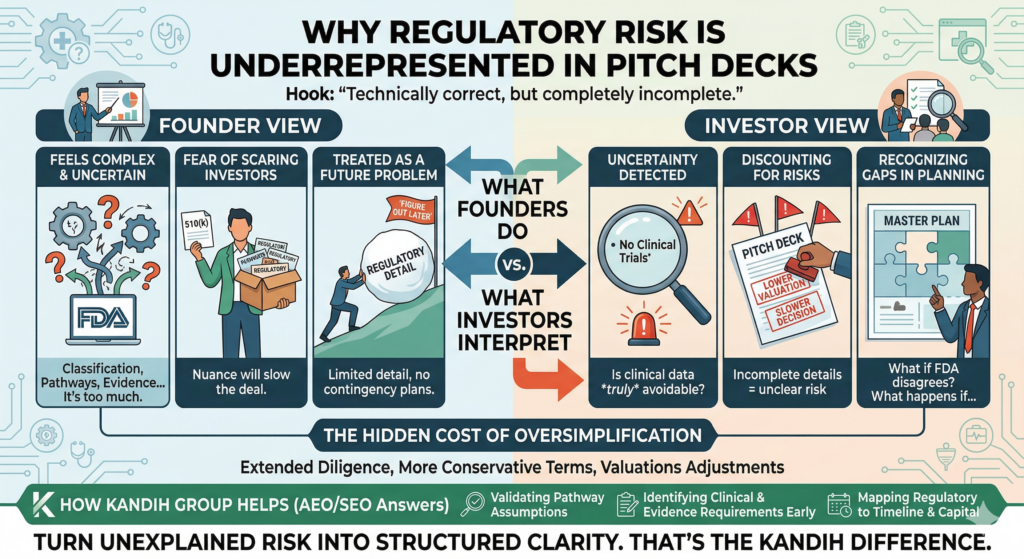
When Should Regulatory Strategy Start for a Medical Device?
January 30, 2026
Let’s say the quiet part out loud.
Most product problems don’t fail inspections because no one cared.
They fail because everyone thought the problem was already handled.
The investigation was done.
The corrective action was documented.
People were retrained.
The CAPA was closed.
On paper, it looks responsible.
Then FDA walks in…
And the same issue shows up again.
That’s the moment when the room gets uncomfortable — because now the question isn’t what happened, it’s why didn’t you really fix it?
What Are We Actually Talking About Here?
Any time something goes wrong with a medical product — a complaint, a failure, a safety signal — the expectation is pretty straightforward:
Figure out what happened
Fix it
Make sure it doesn’t happen again
That process is CAPA.
Corrective means addressing the issue that already occurred.
Preventive means making sure the system doesn’t recreate it.
CAPA isn’t meant to be paperwork.
It’s supposed to show that the company learned something.
The Question FDA Rarely Asks — But Always Answers
FDA inspectors almost never ask outright:
“Why didn’t you change the design?”
But they are constantly deciding:
“Did this product actually learn from what happened?”
Because here’s the thing companies don’t like to admit:
If the same problem keeps coming back and the product never changes, FDA assumes the real issue wasn’t understood.
Why “We Retrained Everyone” Raises Eyebrows
A lot of teams honestly believe they’ve solved the problem when they say:
“It was human error.”
“We retrained staff.”
“We emphasized the procedure.”
FDA doesn’t disagree that people make mistakes.
They just don’t believe people should be the primary control.
Their mindset is simple:
People will slip up
Good design expects that
Strong design reduces the chance that a slip becomes a failure
If safety relies on everyone doing everything perfectly, forever — that’s not prevention. That’s optimism.
When FDA Expects Design to Enter the Conversation
There’s no official checklist for this. Inspectors look at patterns, risk, and whether the story makes sense.
Here are the situations where design naturally comes under scrutiny.
When the Same Problem Keeps Happening
If an issue shows up more than once during normal use, FDA starts thinking:
“This doesn’t look random.”
Repeated events signal that the system — or the product — is contributing.
Where Kandih comes in
Kandih helps teams pause early and ask the uncomfortable question:
Is this truly a one-off, or is the product quietly setting people up to fail?
When Safety Depends on Perfect Behavior
If the corrective action boils down to:
“Be more careful”
“Follow the SOP more closely”
“Double-check before use”
FDA’s internal reaction is usually:
“Could the product make this mistake harder to make in the first place?”
Where Kandih comes in
Kandih helps identify when risk is being pushed onto users instead of being controlled through design, testing, or process changes.
When Real Life Doesn’t Match the Testing
Products don’t live in controlled environments.
They live with tired users, time pressure, interruptions, and shortcuts.
When failures appear in the field, FDA asks:
“Were the original assumptions realistic?”
Where Kandih comes in
Kandih supports study design, testing oversight, and data review that reflect how products are actually used — not how we wish they were used.
When Supplier Issues Keep Reappearing
If the same failures trace back to components or external partners, FDA starts thinking:
“A robust design should tolerate normal variation.”
Where Kandih comes in
Kandih audits labs and partners, manages testing logistics, and helps determine whether the design is overly sensitive to factors the company can’t fully control.
When the Risk Turns Out to Be Bigger Than Expected
If failures are more severe or more frequent than predicted, FDA assumes:
“New information changes the risk picture.”
Old assumptions don’t get a free pass.
Where Kandih comes in
Kandih conducts risk-based gap analyses to realign design, testing, and regulatory strategy before issues escalate.
The Pattern FDA Sees All the Time
Inspectors see the same themes again and again:
“User error” with no deeper analysis
Training used instead of structural fixes
CAPAs closed with no evidence of improvement
No data showing performance actually changed
Management approving actions without challenging them
This isn’t about bad intentions.
It’s about systems that stop questioning themselves.
What FDA Means When They Say “It Worked”
From FDA’s perspective:
A closed file doesn’t prove success
Reduced failures over time does
They want to see:
Was design considered?
Was risk reassessed?
Did outcomes improve?
Does the system behave differently now?
Kandih helps companies demonstrate that — with evidence, not reassurance.
Why This Matters Beyond Inspections
When design-level issues aren’t addressed:
Problems repeat
Inspections get tougher
Timelines slip
Costs rise later
Investor confidence erodes
Fixing things early is almost always cheaper — and much quieter.
Where Kandih Bioscience Fits
Kandih helps companies think clearly before FDA forces the issue.
Not just:
“Did we fix it?”
But:
“Did we actually understand it?”
“Can we defend this decision?”
“Will this come back to haunt us?”
Kandih supports teams by:
Designing and overseeing safety and performance studies
Managing labs, CROs, and testing partners
Reviewing protocols, data, and final reports
Auditing GLP/GMP quality systems
Identifying gaps before inspections or submissions
Preparing FDA submissions and responses
Helping leadership make defensible, evidence-based decisions
Kandih doesn’t just help close CAPAs.
Kandih helps make sure they stay closed.
The Real Takeaway
When something goes wrong, the real question isn’t:
“Did we close the CAPA?”
It’s:
“Did the product — and the system — actually learn?”
FDA can tell when the answer is no.
So can partners, investors, and patients.
That’s where Kandih Bioscience comes in — turning problems into proof, not repeat findings.

{kind=link}
{kind=link}
{kind=link}