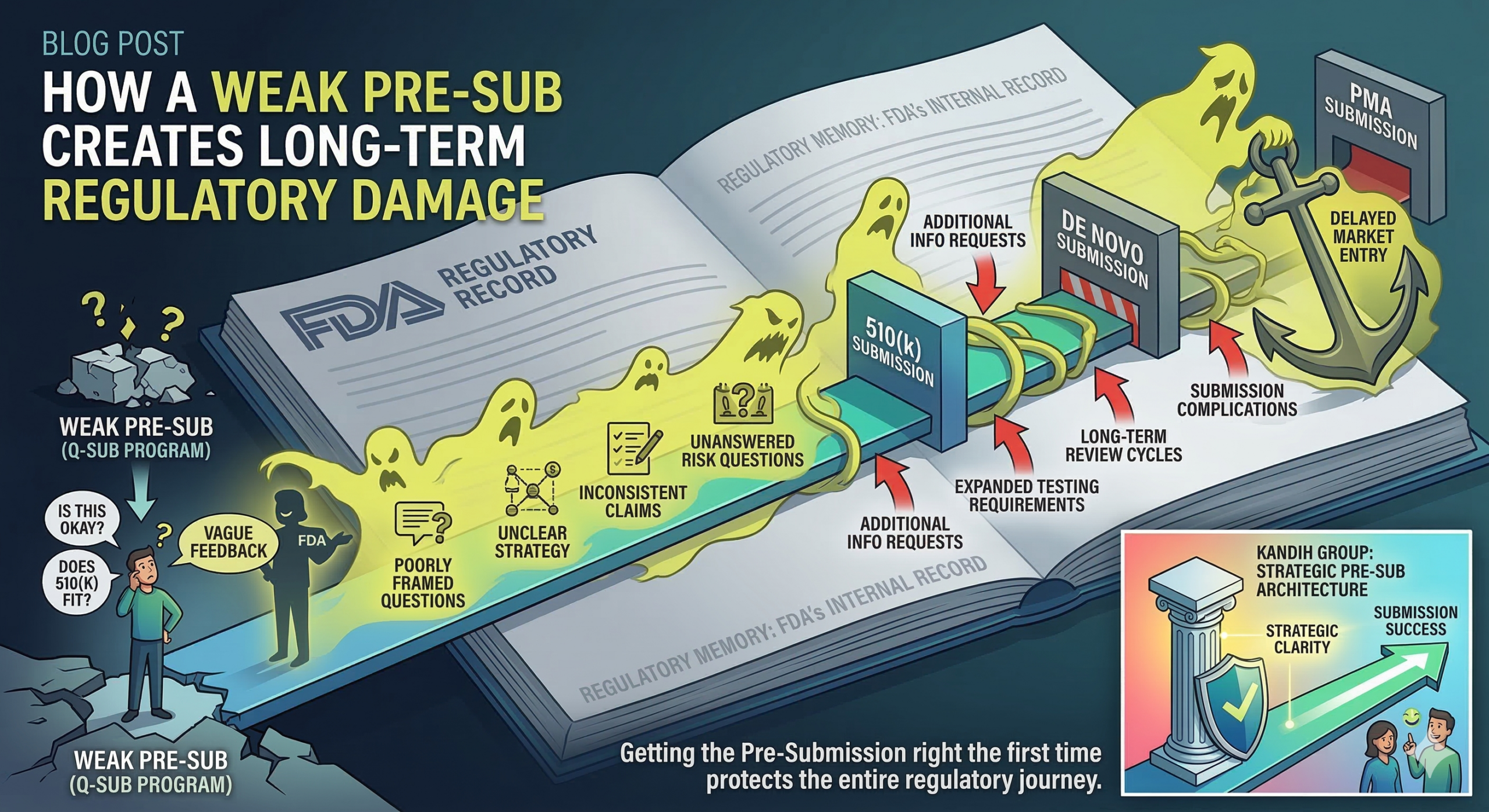
Short answer: a weak Pre-Submission (Pre-Sub) does not just waste a meeting with the FDA. It creates a regulatory record that can follow your device through the rest of development.
Many founders treat a Pre-Sub as a casual conversation with the U.S. Food and Drug Administration. In reality, it is a formal interaction within the FDA’s Q-Submission program. The discussion becomes part of the agency’s internal regulatory history for your device.
That history matters.
When a company later submits a 510(k), De Novo request, or PMA, FDA reviewers can revisit earlier feedback and evaluate whether the company addressed the issues raised.
A poorly prepared Pre-Sub can create problems that persist long after the meeting ends.
Why Pre-Submissions Create a Regulatory Record
FDA documents Pre-Submission interactions internally. These records include:
The briefing package submitted by the company
The questions asked during the meeting
FDA’s written feedback
Internal reviewer notes and risk concerns
When the company later submits a regulatory application, reviewers often reference these earlier discussions to understand the development history.
If early assumptions were weak or poorly framed, those issues can resurface.
This is sometimes called regulatory memory.
How Weak Pre-Subs Cause Long-Term Problems
1. Locking in Poorly Framed Questions
If a company asks vague or poorly structured questions, FDA feedback may also be vague.
Examples include:
“Does our testing plan look reasonable?”
“Do you think 510(k) is appropriate?”
Without clear context, FDA responses may not provide actionable guidance. Later, when problems appear, the company cannot point to clear alignment.
Precise questions produce useful feedback. Weak questions create ambiguity.
2. Signaling Unclear Regulatory Strategy
During a Pre-Sub meeting, FDA reviewers quickly notice if a company:
Has not finalized intended use
Has not validated a predicate strategy
Has not aligned testing with risk analysis
This can create early concern about the company’s regulatory discipline.
Even if the program improves later, those early impressions can influence how reviewers approach the submission.
3. Introducing Inconsistent Claims
Another common issue occurs when:
Intended use language evolves after the Pre-Sub
Claims expand during development
Testing strategies shift without explanation
If the final submission diverges from earlier discussions without clear justification, FDA may question the rationale.
Consistency matters.
4. Creating a Record of Unanswered Risk Questions
If the Pre-Sub identifies safety questions that are not fully addressed later, reviewers may return to those concerns during submission review.
This can trigger:
Additional information requests
Expanded testing requirements
Longer review cycles
In other words, unresolved issues can carry forward.
AEO: Common Questions About FDA Pre-Submissions
Does an FDA Pre-Submission create a regulatory record?
Yes. Pre-Sub interactions become part of the FDA’s internal regulatory history for the device.
Can a poorly prepared Pre-Sub affect later submissions?
Yes. Weak questions or unclear strategy may lead to ambiguous feedback that complicates later regulatory interactions.
How can companies avoid Pre-Sub mistakes?
By clearly defining intended use, regulatory pathway logic, and evidence strategy before engaging FDA.
Where Kandih Comes In
This is where Kandih Group helps companies structure Pre-Submissions strategically.
Kandih approaches Pre-Subs as part of a long-term regulatory architecture.
We help teams:
Clarify intended use before FDA interaction
Validate pathway assumptions
Frame device risks clearly
Align testing strategies with regulatory expectations
Draft focused questions that produce actionable feedback
Ensure consistency between Pre-Sub discussions and future submissions
The goal is simple: create a regulatory record that strengthens the program instead of complicating it.
A well-structured Pre-Sub becomes an anchor for the entire development plan.
Bottom Line
Pre-Submissions are not informal conversations.
They establish early regulatory alignment and create a documented interaction with FDA reviewers.
Weak Pre-Subs create ambiguity.
Strong Pre-Subs create strategic clarity.
When done properly, they reduce downstream surprises and improve submission stability.
Getting the Pre-Submission right the first time protects the entire regulatory journey.
References
FDA – Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/requests-feedback-and-meetings-medical-device-submissions-q-submission-program
FDA – Premarket Notification 510(k)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-notification-510k
FDA – De Novo Classification Process
https://www.fda.gov/medical-devices/premarket-submissions/de-novo-classification-request
FDA – Design Controls for Medical Devices
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/design-controls-medical-devices