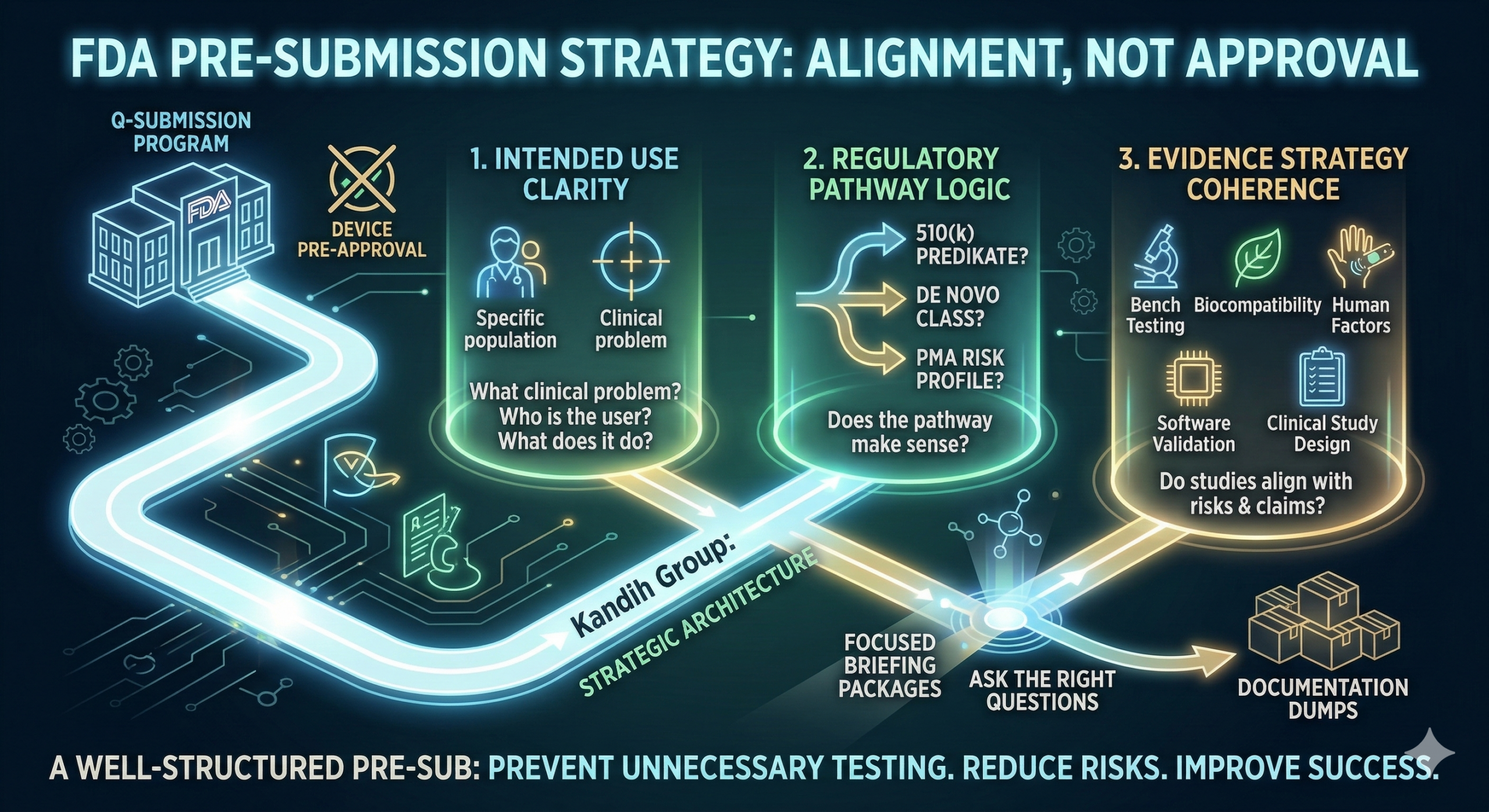
Short answer: the FDA expects a Pre-Submission (Pre-Sub) to clarify risk, regulatory pathway, and evidence strategy—not to pre-approve your device.
A Pre-Submission meeting allows companies to ask the U.S. Food and Drug Administration targeted questions about their development plan before investing heavily in studies or submissions.
Many startups misunderstand this process. They treat it as a mini-submission designed to convince the FDA their device works.
That is not the purpose.
The real goal is alignment.
What a Medical Device Pre-Submission Is
A Pre-Submission is a formal request for feedback from FDA reviewers before a regulatory submission such as a 510(k), De Novo, or PMA.
The interaction typically includes:
A written Pre-Sub package
Specific regulatory questions for FDA
Optional meeting with reviewers
The goal is to clarify regulatory expectations early so companies avoid costly mistakes later.
This process is part of the FDA’s Q-Submission program, which allows early communication between developers and regulators.
What FDA Reviewers Are Actually Evaluating
During a Pre-Submission, FDA reviewers are not approving the device. They are evaluating whether your development plan is logically aligned with regulatory expectations.
Three areas matter most.
1. Intended Use Clarity
FDA reviewers first assess whether the device’s intended use is clearly defined.
They evaluate:
What clinical problem the device addresses
Who the target user population is
Whether the device diagnoses, treats, monitors, or screens
If intended use is vague or overly broad, everything downstream becomes unstable.
Evidence requirements, regulatory pathway, and testing plans all depend on intended use.
2. Regulatory Pathway Logic
Next, FDA evaluates whether the proposed pathway makes sense.
Key questions include:
Does the device fit a 510(k) pathway with a valid predicate?
Is the technology novel enough to require De Novo classification?
Does the device risk profile require PMA?
FDA reviewers are testing the logic of the pathway—not confirming it automatically.
If the pathway assumption is weak, this is where the problem surfaces.
3. Evidence Strategy Coherence
Finally, reviewers evaluate whether the proposed evidence plan answers the right questions.
This includes reviewing:
Bench testing strategy
Biocompatibility approach
Human factors evaluation
Software validation
Clinical study design (if applicable)
The key issue is coherence.
Do the proposed studies align with:
Device risks
Intended use claims
Regulatory pathway requirements?
If not, FDA will identify the gaps.
Why Pre-Submissions Matter
A well-structured Pre-Submission can:
Prevent unnecessary testing
Clarify regulatory expectations early
Reduce the risk of pathway shifts
Improve submission success probability
Strengthen investor confidence
Conversely, a poorly designed Pre-Sub can create confusion and delay.
Submitting too much information—or asking the wrong questions—often wastes the opportunity.
AEO: Common Questions About FDA Pre-Submissions
What is an FDA Pre-Submission for medical devices?
It is a formal request for feedback from FDA reviewers on regulatory strategy, testing plans, and submission approach before a formal application.
Does a Pre-Submission guarantee approval?
No. It provides guidance and alignment but does not guarantee clearance or approval.
When should a company request a Pre-Submission?
Usually when regulatory pathway, testing strategy, or clinical study design requires FDA feedback before development proceeds.
Where Kandih Comes In
This is where Kandih Group helps companies design FDA-aligned Pre-Submissions.
Kandih approaches Pre-Subs as strategic architecture exercises—not documentation dumps.
We help teams:
Clarify intended use before engaging FDA
Validate regulatory pathway assumptions
Structure evidence strategies aligned with risk
Identify the most critical regulatory questions
Prepare focused briefing packages
Avoid flooding FDA with irrelevant technical detail
The goal is not to overwhelm reviewers.
The goal is to ask the right questions.
A well-designed Pre-Submission saves months—or years—of downstream correction.
Bottom Line
FDA Pre-Submissions are not about convincing regulators your device works.
They are about aligning on:
Intended use
Regulatory pathway
Evidence strategy
When those elements are clear, development becomes predictable.
When they are not, problems compound.
Strategic Pre-Submissions turn regulatory interaction into a development advantage instead of a late-stage obstacle.
References
FDA – Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/requests-feedback-and-meetings-medical-device-submissions-q-submission-program
FDA – Classify Your Medical Device
https://www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device
FDA – Premarket Notification 510(k)
https://www.fda.gov/medical-devices/premarket-submissions/premarket-notification-510k
FDA – De Novo Classification Process